 W
WAceDB is a biological database for handling genomic data. It was developed by Richard M. Durbin and Jean Thierry-Mieg in 1989. AceDB stands for a C. elegans database. Although AceDB was initially created as a database specifically for the nematode worm it has also come to mean the database software itself, which has been used to store information for other species. According to its website, AceDB provides a custom database kernel with a non-standard data model designed with flexibility in mind. For example, there is an AceDB instance for the organism Pristionchus pacificus called AppaDB. Much of the functionality of AceDB for C. elegans has been made available through the WormBase database.
 W
WThe Actino-ugpB RNA motif is a conserved RNA structure that was discovered by bioinformatics. Actino-ugpB motifs are found in strains of the species Gardnerella vaginalis, within the phylum Actinobacteria.
The Bioinformatics Open Source Conference (BOSC) is an academic conference on open-source programming in bioinformatics organised by the Open Bioinformatics Foundation. The conference has been held annually since 2000 and is run as a two-day satellite meeting preceding the Intelligent Systems for Molecular Biology (ISMB) conference.
Blast2GO, first published in 2005, is a bioinformatics software tool for the automatic, high-throughput functional annotation of novel sequence data. It makes use of the BLAST algorithm to identify similar sequences to then transfers existing functional annotation from yet characterised sequences to the novel one. The functional information is represented via the Gene Ontology (GO), a controlled vocabulary of functional attributes. The Gene Ontology, or GO, is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
 W
WBottom-up proteomics is a common method to identify proteins and characterize their amino acid sequences and post-translational modifications by proteolytic digestion of proteins prior to analysis by mass spectrometry. The major alternative workflow used in proteomics is called top-down proteomics where intact proteins are purified prior to digestion and/or fragmentation either within the mass spectrometer or by 2D electrophoresis. Essentially, bottom-up proteomics is a relatively simple and reliable means of determining the protein make-up of a given sample of cells, tissues, etc.
 W
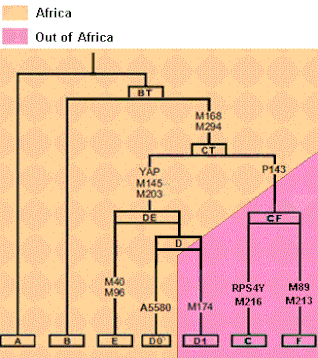
WHaplogroup BT M91, also known as Haplogroup A1b2, is a Y-chromosome haplogroup. BT is a subclade of haplogroup A1b (P108) and a sibling of the haplogroup A1b1 (L419/PF712).
 W
WHaplogroup D-Z27276 also known as Haplogroup D1a1 is a Y-chromosome haplogroup. It is one of two branches of Haplogroup D1, one of the descendants of Haplogroup D. The other is D-M55 which is only found in Japan.
 W
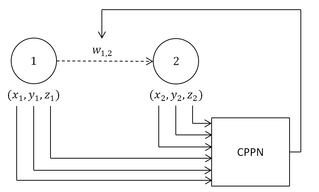
WHypercube-based NEAT, or HyperNEAT, is a generative encoding that evolves artificial neural networks (ANNs) with the principles of the widely used NeuroEvolution of Augmented Topologies (NEAT) algorithm. It is a novel technique for evolving large-scale neural networks using the geometric regularities of the task domain. It uses Compositional Pattern Producing Networks (CPPNs), which are used to generate the images for Picbreeder.org and shapes for EndlessForms.com. HyperNEAT has recently been extended to also evolve plastic ANNs and to evolve the location of every neuron in the network.
 W
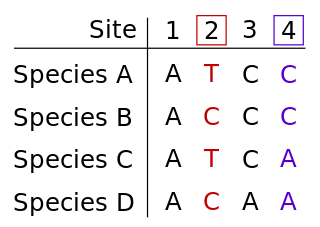
WIn phylogenetics, informative site is a term used in the context of maximum parsimony, to refer to a characteristic that can usefully distinguish between samples at a genetic level. The informative site is a position in the relevant set of sequences at which there are at least two different character states at that point in the sequences, and each of those states occurs in at least two of the sequences. Character states can take on multiple types of data, including morphological or molecular information such as sequences of DNA or proteins.
 W
WThe Journal of Computational Biology is a monthly peer-reviewed scientific journal covering computational biology and bioinformatics. It was established in 1994 and is published by Mary Ann Liebert, Inc. The editors-in-chief are Sorin Istrail and Michael S. Waterman. According to the Journal Citation Reports, the journal has a 2018 impact factor of 0.879.
 W
WIn bioinformatics, MAFFT is a program used to create multiple sequence alignments of amino acid or nucleotide sequences. Published in 2002, the first version of MAFFT used an algorithm based on progressive alignment, in which the sequences were clustered with the help of the Fast Fourier Transform. Subsequent versions of MAFFT have added other algorithms and modes of operation, including options for faster alignment of large numbers of sequences, higher accuracy alignments, alignment of non-coding RNA sequences, and the addition of new sequences to existing alignments.
 W
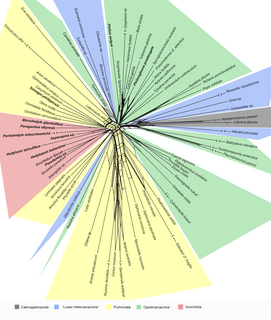
WNeighborNet is an algorithm for constructing phylogenetic networks which is loosely based on the neighbor joining algorithm. Like neighbor joining, the method takes a distance matrix as input, and works by agglomerating clusters. However, the NeighborNet algorithm can lead to collections of clusters which overlap and do not form a hierarchy, and are represented using a type of phylogenetic network called a splits graph. If the distance matrix satisfies the Kalmanson combinatorial conditions then Neighbor-net will return the corresponding circular ordering. The method is implemented in the SplitsTree and R/Phangorn packages.