 W
WMolecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
 W
WThe accessible surface area (ASA) or solvent-accessible surface area (SASA) is the surface area of a biomolecule that is accessible to a solvent. Measurement of ASA is usually described in units of square Ångstroms. ASA was first described by Lee & Richards in 1971 and is sometimes called the Lee-Richards molecular surface. ASA is typically calculated using the 'rolling ball' algorithm developed by Shrake & Rupley in 1973. This algorithm uses a sphere of a particular radius to 'probe' the surface of the molecule.
 W
WThe Anisotropic Network Model (ANM) is a simple yet powerful tool made for Normal Mode Analysis of proteins, which has been successfully applied for exploring the relation between function and dynamics for many proteins. It is essentially an Elastic Network Model for the Cα atoms with a step function for the dependence of the force constants on the inter-particle distance.
 W
WIn chemistry, the ball-and-stick model is a molecular model of a chemical substance which is to display both the three-dimensional position of the atoms and the bonds between them. The atoms are typically represented by spheres, connected by rods which represent the bonds. Double and triple bonds are usually represented by two or three curved rods, respectively, or alternately by correctly positioned sticks for the sigma and pi bonds. In a good model, the angles between the rods should be the same as the angles between the bonds, and the distances between the centers of the spheres should be proportional to the distances between the corresponding atomic nuclei. The chemical element of each atom is often indicated by the sphere's color.
 W
WSpiroligomers are synthetic oligomers made by coupling pairs of bis-amino acids into a fused ring system. Spiroligomers are rich in stereochemistry and functionality because of the variety of bis-amino acids that are capable of being incorporated during synthesis. Due to the rigidity of the fused ring system, the three-dimensional shape of a spiroligomer – as well as the display of any functional groups – can be predicted, allowing for molecular modeling and dynamics.
WIn chemistry, the CPK coloring is a popular color convention for distinguishing atoms of different chemical elements in molecular models. The scheme is named after the CPK molecular models designed by chemists Robert Corey and Linus Pauling, and improved by Walter Koltun.
 W
WA dial box is a computer peripheral for direct 3D manipulation e.g. to interactively input the rotation and torsion angles of a model displayed on a computer screen. Dial boxes were common input tools in the first years of interactive 3D graphics and they were available for Silicon Graphics (SGI) or Sun Microsystems and sold with their workstations. Currently they have been replaced by standard computer mouse interaction techniques.
 W
WIn the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
 W
WIn the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
 W
WThe Gaussian network model (GNM) is a representation of a biological macromolecule as an elastic mass-and-spring network to study, understand, and characterize the mechanical aspects of its long-time large-scale dynamics. The model has a wide range of applications from small proteins such as enzymes composed of a single domain, to large macromolecular assemblies such as a ribosome or a viral capsid. Protein domain dynamics plays key roles in a multitude of molecular recognition and cell signalling processes. Protein domains, connected by intrinsically disordered flexible linker domains, induce long-range allostery via protein domain dynamics. The resultant dynamic modes cannot be generally predicted from static structures of either the entire protein or individual domains.
 W
WMolecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanics force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
WMolecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
WMolecular modeling on GPU is the technique of using a graphics processing unit (GPU) for molecular simulations.
 W
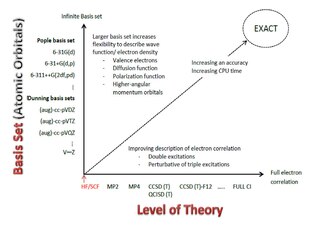
WA Pople diagram or Pople's Diagram is a diagram which describes the relationship between various calculation methods in computational chemistry. It was initially introduced in January 1965 by Sir John Pople,, during the Symposium of Atomic and Molecular Quantum Theory in Florida. The Pople Diagram can be either 2-dimensional or 3-dimensional, with the axes representing ab inito methods, basis sets and treatment of relativity. The diagram attempts to balance calculations by giving all aspects of a computation equal weight.
Rosetta@home is a distributed computing project for protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker laboratory at the University of Washington. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
 W
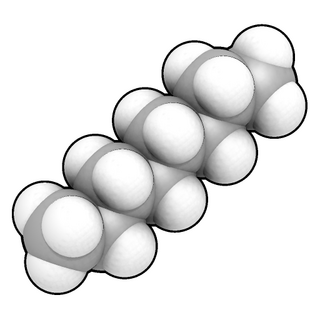
WIn chemistry, a space-filling model, also known as a calotte model, is a type of three-dimensional (3D) molecular model where the atoms are represented by spheres whose radii are proportional to the radii of the atoms and whose center-to-center distances are proportional to the distances between the atomic nuclei, all in the same scale. Atoms of different chemical elements are usually represented by spheres of different colors.
WSpiroligomers are synthetic oligomers made by coupling pairs of bis-amino acids into a fused ring system. Spiroligomers are rich in stereochemistry and functionality because of the variety of bis-amino acids that are capable of being incorporated during synthesis. Due to the rigidity of the fused ring system, the three-dimensional shape of a spiroligomer – as well as the display of any functional groups – can be predicted, allowing for molecular modeling and dynamics.
 W
WIn protein structure prediction, a statistical potential or knowledge-based potential is a scoring function derived from an analysis of known protein structures in the Protein Data Bank (PDB).