 W
WChemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.
 W
WChemical space is a concept in cheminformatics referring to the property space spanned by all possible molecules and chemical compounds adhering to a given set of construction principles and boundary conditions. It contains millions of compounds which are readily accessible and available to researchers. It is a library used in the method of molecular docking.
 W
WChemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
 W
WDynamic combinatorial chemistry (DCC); also known as constitutional dynamic chemistry (CDC) is a method to the generation of new molecules formed by reversible reaction of simple building blocks under thermodynamic control. The library of these reversibly interconverting building blocks is called a dynamic combinatorial library (DCL). All constituents in a DCL are in equilibrium, and their distribution is determined by their thermodynamic stability within the DCL. The interconversion of these building blocks may involve covalent or non-covalent interactions. When a DCL is exposed to an external influence, the equilibrium shifts and those components that interact with the external influence are stabilised and amplified, allowing more of the active compound to be formed.
 W
WThe Hosoya index, also known as the Z index, of a graph is the total number of matchings in it. The Hosoya index is always at least one, because the empty set of edges is counted as a matching for this purpose. Equivalently, the Hosoya index is the number of non-empty matchings plus one. The index is named after Haruo Hosoya.
 W
WThe Journal of Chemical Information and Modeling is a peer-reviewed scientific journal published by the American Chemical Society. It was established in 1961 as the Journal of Chemical Documentation, renamed in 1975 to Journal of Chemical Information and Computer Sciences, and obtained its current name in 2005. The journal covers the fields of chemical informatics and molecular modeling. The editor-in-chief is Kenneth M. Merz Jr.. The journal supports Open Science approaches.
 W
WThe Journal of Cheminformatics is a peer-reviewed open access scientific journal that covers cheminformatics and molecular modelling. It was established in 2009 with David Wild and Christoph Steinbeck as founding editors-in-chief, and was originally published by Chemistry Central. At the end of 2015, the Chemistry Central brand was retired and its titles, including Journal of Cheminformatics, were merged with the SpringerOpen portfolio of open access journals.
 W
WLipinski's rule of five, also known as Pfizer's rule of five or simply the rule of five (RO5), is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has chemical properties and physical properties that would make it a likely orally active drug in humans. The rule was formulated by Christopher A. Lipinski in 1997, based on the observation that most orally administered drugs are relatively small and moderately lipophilic molecules.
 W
WMedicinal chemistry and pharmaceutical chemistry are disciplines at the intersection of chemistry, especially synthetic organic chemistry, and pharmacology and various other biological specialties, where they are involved with design, chemical synthesis and development for market of pharmaceutical agents, or bio-active molecules (drugs).
 W
WIn chemical graph theory and in mathematical chemistry, a molecular graph or chemical graph is a representation of the structural formula of a chemical compound in terms of graph theory. A chemical graph is a labeled graph whose vertices correspond to the atoms of the compound and edges correspond to chemical bonds. Its vertices are labeled with the kinds of the corresponding atoms and edges are labeled with the types of bonds. For particular purposes any of the labelings may be ignored.
 W
WA Savitzky–Golay filter is a digital filter that can be applied to a set of digital data points for the purpose of smoothing the data, that is, to increase the precision of the data without distorting the signal tendency. This is achieved, in a process known as convolution, by fitting successive sub-sets of adjacent data points with a low-degree polynomial by the method of linear least squares. When the data points are equally spaced, an analytical solution to the least-squares equations can be found, in the form of a single set of "convolution coefficients" that can be applied to all data sub-sets, to give estimates of the smoothed signal, at the central point of each sub-set. The method, based on established mathematical procedures, was popularized by Abraham Savitzky and Marcel J. E. Golay, who published tables of convolution coefficients for various polynomials and sub-set sizes in 1964. Some errors in the tables have been corrected. The method has been extended for the treatment of 2- and 3-dimensional data.
 W
WA pharmacophore is an abstract description of molecular features that are necessary for molecular recognition of a ligand by a biological macromolecule. IUPAC defines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger its biological response". A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. Furthermore, pharmacophore models can be used to identify through de novo design or virtual screening novel ligands that will bind to the same receptor.
 W
WThe polar surface area (PSA) or topological polar surface area (TPSA) of a molecule is defined as the surface sum over all polar atoms or molecules, primarily oxygen and nitrogen, also including their attached hydrogen atoms.
 W
WSensitivity and specificity are statistical measures of the performance of a binary classification test that are widely used in medicine:Sensitivity measures the proportion of positives that are correctly identified. Specificity measures the proportion of negatives that are correctly identified.
 W
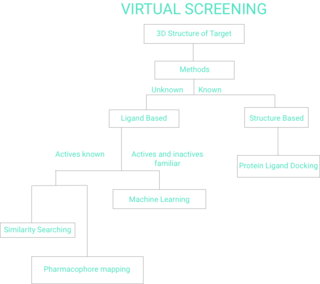
WVirtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
 W
W W
W W
W W
W W
W W
W W
W W
W W
W W
W