 W
WThe accessible surface area (ASA) or solvent-accessible surface area (SASA) is the surface area of a biomolecule that is accessible to a solvent. Measurement of ASA is usually described in units of square Ångstroms. ASA was first described by Lee & Richards in 1971 and is sometimes called the Lee-Richards molecular surface. ASA is typically calculated using the 'rolling ball' algorithm developed by Shrake & Rupley in 1973. This algorithm uses a sphere of a particular radius to 'probe' the surface of the molecule.
 W
WBond order potential is a class of empirical (analytical) interatomic potentials which is used in molecular dynamics and molecular statics simulations. Examples include the Tersoff potential, the EDIP potential, the Brenner potential, the Finnis-Sinclair potentials, ReaxFF, and the second-moment tight-binding potentials. They have the advantage over conventional molecular mechanics force fields in that they can, with the same parameters, describe several different bonding states of an atom, and thus to some extent may be able to describe chemical reactions correctly. The potentials were developed partly independently of each other, but share the common idea that the strength of a chemical bond depends on the bonding environment, including the number of bonds and possibly also angles and bond lengths. It is based on the Linus Pauling bond order concept and can be written in the form
 W
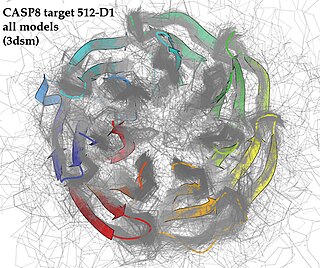
WCritical Assessment of protein Structure Prediction, or CASP, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence, many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.
 W
WThe Collaborative Computational Projects (CCP) group was responsible for the development of CCPForge, which is a software development tool produced through collaborations by the CCP community. CCPs allow experts in computational research to come together and develop scientific software which can be applied to numerous research fields. It is used as a tool in many research and development areas, and hosts a variety of projects. Every CCP project is the result of years of valuable work by computational researchers.
 W
WChemical space is a concept in cheminformatics referring to the property space spanned by all possible molecules and chemical compounds adhering to a given set of construction principles and boundary conditions. It contains millions of compounds which are readily accessible and available to researchers. It is a library used in the method of molecular docking.
 W
WChemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
 W
WCOSMO is the abbreviation for "COnductor-like Screening MOdel", a calculation method for determining the electrostatic interaction of a molecule with a solvent. The method is commonly used in computational chemistry to model solvation effects.
 W
WIn the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
 W
WA quantum mechanical system or particle that is bound—that is, confined spatially—can only take on certain discrete values of energy, called energy levels. This contrasts with classical particles, which can have any amount of energy. The term is commonly used for the energy levels of the electrons in atoms, ions, or molecules, which are bound by the electric field of the nucleus, but can also refer to energy levels of nuclei or vibrational or rotational energy levels in molecules. The energy spectrum of a system with such discrete energy levels is said to be quantized.(And the energy levels don’t have to be equal )
 W
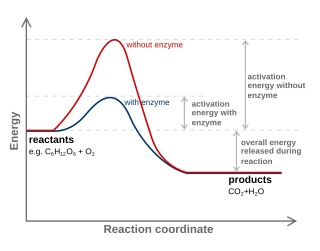
WFor a chemical reaction or process an energy profile is a theoretical representation of a single energetic pathway, along the reaction coordinate, as the reactants are transformed into products. Reaction coordinate diagrams are derived from the corresponding potential energy surface (PES), which are used in computational chemistry to model chemical reactions by relating the energy of a molecule(s) to its structure. The reaction coordinate is a parametric curve that follows the pathway of a reaction and indicates the progress of a reaction.
 W
WThe International Journal of Quantum Chemistry is a peer-reviewed scientific journal publishing original, primary research and review articles on all aspects of quantum chemistry, including an expanded scope focusing on aspects of materials science, biochemistry, biophysics, quantum physics, quantum information theory, etc.
 W
WThe Journal of Chemical Information and Modeling is a peer-reviewed scientific journal published by the American Chemical Society. It was established in 1961 as the Journal of Chemical Documentation, renamed in 1975 to Journal of Chemical Information and Computer Sciences, and obtained its current name in 2005. The journal covers the fields of chemical informatics and molecular modeling. The editor-in-chief is Kenneth M. Merz Jr.. The journal supports Open Science approaches.
 W
WJournal of Chemical Theory and Computation is a peer-reviewed scientific journal, established in 2005 by the American Chemical Society. It is indexed in Chemical Abstracts Service (CAS), Scopus, British Library, and Web of Science. The current editors are William L. Jorgensen and Gustavo E. Scuseria. Currently as of the year 2015, JCTC has 11 volumes.
 W
WBette Korber is an American computational biologist focusing on the molecular biology and population genetics of the HIV virus that causes infection and eventually AIDS. She has contributed heavily to efforts to obtain an effective HIV vaccine. She created a database at Los Alamos National Laboratory that has enabled her to design novel mosaic HIV vaccines, one of which is currently in human testing in Africa. The database contains thousands of HIV genome sequences and related data.
 W
WThe Lennard-Jones potential is an intermolecular pair potential. Among the intermolecular potentials, the Lennard-Jones potential has a central role as water among real fluids: It is the potential that has been studied most extensively and most thoroughly. It is considered as archetype model for simple yet realistic intermolecular interactions.
 W
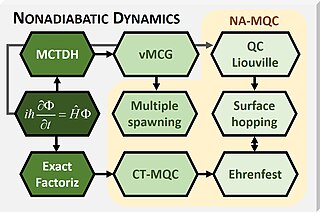
WMixed quantum-classical (MQC) dynamics is a class of computational theoretical chemistry methods tailored to simulate nonadiabatic (NA) processes in molecular and supramolecular chemistry. Such methods are characterized by:Propagation of nuclear dynamics through classical trajectories; Propagation of the electrons through quantum methods; A feedback algorithm between the electronic and nuclear subsystems to recover nonadiabatic information.
 W
WMolecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanics force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
 W
WMolecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
 W
WMolecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
WMolecular modeling on GPU is the technique of using a graphics processing unit (GPU) for molecular simulations.
 W
WIn chemistry, a molecular orbital is a mathematical function describing the location and wave-like behavior of an electron in a molecule. This function can be used to calculate chemical and physical properties such as the probability of finding an electron in any specific region. The term orbital was introduced by Robert S. Mulliken in 1932 as an abbreviation for one-electron orbital wave function. At an elementary level, it is used to describe the region of space in which the function has a significant amplitude. In an isolated atom, the orbital electrons' location is determined by functions called atomic orbitals. When multiple atoms combine chemically into a molecule, the electrons' locations are determined by the molecule as a whole, so the atomic orbitals combine to form molecular orbitals. The electrons from the constituent atoms occupy the molecular orbitals. Mathematically, molecular orbitals are an approximate solution to the Schrodinger equation for the electrons in the field of the molecule's atomic nuclei. They are usually constructed by combining atomic orbitals or hybrid orbitals from each atom of the molecule, or other molecular orbitals from groups of atoms. They can be quantitatively calculated using the Hartree–Fock or self-consistent field (SCF) methods.
 W
WIn atomic physics and quantum chemistry, the electron configuration is the distribution of electrons of an atom or molecule in atomic or molecular orbitals. For example, the electron configuration of the neon atom is 1s2 2s2 2p6, using the notation explained below.
 W
WIn mathematics and physics, perturbation theory comprises mathematical methods for finding an approximate solution to a problem, by starting from the exact solution of a related, simpler problem. A critical feature of the technique is a middle step that breaks the problem into "solvable" and "perturbative" parts. Perturbation theory is widely used when the problem at hand does not have a known exact solution, but can be expressed as a "small" change to a known solvable problem. Perturbation theory is used in a wide range of fields, and reaches its most sophisticated and advanced forms in quantum field theory. Perturbation theory for quantum mechanics imparts the first step on this path. The field in general remains actively and heavily researched across multiple disciplines.
 W
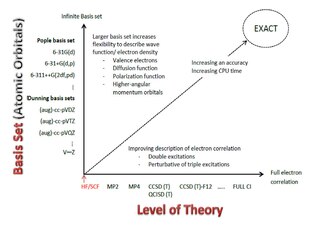
WA Pople diagram or Pople's Diagram is a diagram which describes the relationship between various calculation methods in computational chemistry. It was initially introduced in January 1965 by Sir John Pople,, during the Symposium of Atomic and Molecular Quantum Theory in Florida. The Pople Diagram can be either 2-dimensional or 3-dimensional, with the axes representing ab inito methods, basis sets and treatment of relativity. The diagram attempts to balance calculations by giving all aspects of a computation equal weight.
 W
WIn chemistry, a reaction coordinate is an abstract one-dimensional coordinate which represents progress along a reaction pathway. It is usually a geometric parameter that changes during the conversion of one or more molecular entities. In molecular dynamics simulations, a reaction coordinate is called collective variable.
 W
WIn mathematics, a self-avoiding walk (SAW) is a sequence of moves on a lattice that does not visit the same point more than once. This is a special case of the graph theoretical notion of a path. A self-avoiding polygon (SAP) is a closed self-avoiding walk on a lattice. Very little is known rigorously about the self-avoiding walk from a mathematical perspective, although physicists have provided numerous conjectures that are believed to be true and are strongly supported by numerical simulations.
 W
WUmbrella sampling is a technique in computational physics and chemistry, used to improve sampling of a system where ergodicity is hindered by the form of the system's energy landscape. It was first suggested by Torrie and Valleau in 1977. It is a particular physical application of the more general importance sampling in statistics.
 W
WIn computational chemistry, a water model is used to simulate and thermodynamically calculate water clusters, liquid water, and aqueous solutions with explicit solvent. The models are determined from quantum mechanics, molecular mechanics, experimental results, and these combinations. To imitate a specific nature of molecules, many types of models have been developed. In general, these can be classified by the following three points; (i) the number of interaction points called site, (ii) whether the model is rigid or flexible, (iii) whether the model includes polarization effects.
 W
WZanamivir is a medication used to treat and prevent influenza caused by influenza A and B viruses. It is a neuraminidase inhibitor and was developed by the Australian biotech firm Biota Holdings. It was licensed to Glaxo in 1990 and approved in the US in 1999, only for use as a treatment for influenza. In 2006, it was approved for prevention of influenza A and B. Zanamivir was the first neuraminidase inhibitor commercially developed. It is currently marketed by GlaxoSmithKline under the trade name Relenza as a powder for oral inhalation.