Anduril is an open source component-based workflow framework for scientific data analysis developed at the Systems Biology Laboratory, University of Helsinki.
Apache cTAKES: clinical Text Analysis and Knowledge Extraction System is an open-source Natural Language Processing (NLP) system that extracts clinical information from electronic health record unstructured text. It processes clinical notes, identifying types of clinical named entities — drugs, diseases/disorders, signs/symptoms, anatomical sites and procedures. Each named entity has attributes for the text span, the ontology mapping code, context, and negated/not negated.
Apache Taverna is an open source software tool for designing and executing workflows, initially created by the myGrid project under the name Taverna Workbench, now a project under the Apache incubator. Taverna allows users to integrate many different software components, including WSDL SOAP or REST Web services, such as those provided by the National Center for Biotechnology Information, the European Bioinformatics Institute, the DNA Databank of Japan (DDBJ), SoapLab, BioMOBY and EMBOSS. The set of available services is not finite and users can import new service descriptions into the Taverna Workbench.
 W
WArrayTrack is a multi-purpose bioinformatics tool primarily used for microarray data management, analysis, and interpretation. ArrayTrack was developed to support in-house filter array research for the U.S. Food and Drug Administration in 2001 and was made freely available to the public as an integrated research tool for microarrays in 2003. Since then, ArrayTrack has averaged about 5,000 users per year. It is regularly updated by the National Center for Toxicological Research.
 W
WBioMart is a community-driven project to provide a single point of access to distributed research data. The BioMart project contributes open source software and data services to the international scientific community. Although the BioMart software is primarily used by the biomedical research community, it is designed in such a way that any type of data can be incorporated into the BioMart framework. The BioMart project originated at the European Bioinformatics Institute as a data management solution for the Human Genome Project. Since then, BioMart has grown to become a multi-institute collaboration involving various database projects on five continents.
 W
WBioNumerics is a suite of bioinformatics software applications developed by the company Applied Maths NV.
 W
WBioPerl is a collection of Perl modules that facilitate the development of Perl scripts for bioinformatics applications. It has played an integral role in the Human Genome Project.
The Biopython project is an open-source collection of non-commercial Python tools for computational biology and bioinformatics, created by an international association of developers. It contains classes to represent biological sequences and sequence annotations, and it is able to read and write to a variety of file formats. It also allows for a programmatic means of accessing online databases of biological information, such as those at NCBI. Separate modules extend Biopython's capabilities to sequence alignment, protein structure, population genetics, phylogenetics, sequence motifs, and machine learning. Biopython is one of a number of Bio* projects designed to reduce code duplication in computational biology.
BioRuby is a collection of open-source Ruby code, comprising classes for computational molecular biology and bioinformatics. It contains classes for DNA and protein sequence analysis, sequence alignment, biological database parsing, structural biology and other bioinformatics tasks.
 W
WBioSLAX is a Live CD/Live DVD/Live USB comprising a suite of more than 300 bioinformatics tools and application suites. It has been released by the Bioinformatics Resource Unit of the Life Sciences Institute (LSI), National University of Singapore (NUS) and is bootable from any PC that allows a CD/DVD or USB boot option and runs the compressed Slackware flavour of the Linux Operating System (OS), also known as Slax. Slax was created by Tomáš Matějíček in the Czech Republic using the Linux Live Scripts which he also developed. The BioSLAX derivative was created by Mark De Silva, Lim Kuan Siong and Tan Tin Wee.
Blast2GO, first published in 2005, is a bioinformatics software tool for the automatic, high-throughput functional annotation of novel sequence data. It makes use of the BLAST algorithm to identify similar sequences to then transfers existing functional annotation from yet characterised sequences to the novel one. The functional information is represented via the Gene Ontology (GO), a controlled vocabulary of functional attributes. The Gene Ontology, or GO, is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
 W
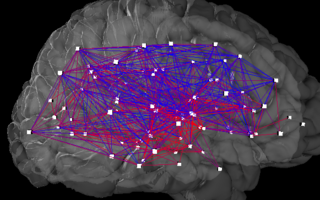
WThe Budapest Reference Connectome server computes the frequently appearing anatomical brain connections of 418 healthy subjects. It has been prepared from diffusion MRI datasets of the Human Connectome Project into a reference connectome, which can be downloaded in CSV and GraphML formats and visualized on the site in 3D.
 W
WThe Chemistry Development Kit (CDK) is computer software, a library in the programming language Java, for chemoinformatics and bioinformatics. It is available for Windows, Linux, Unix, and macOS. It is free and open-source software distributed under the GNU Lesser General Public License (LGPL) 2.0.
 W
WCheShift-2 is an application created to compute 13Cα and 13Cβ protein chemical shifts and to validate protein structures. It is based on quantum mechanics computations of 13Cα and 13Cβchemical shift as a function of the torsional angles of the 20 amino acids.
 W
WClustal is a series of widely used computer programs used in Bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm are also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
 W
WCytoscape is an open source bioinformatics software platform for visualizing molecular interaction networks and integrating with gene expression profiles and other state data. Additional features are available as plugins. Plugins are available for network and molecular profiling analyses, new layouts, additional file format support and connection with databases and searching in large networks. Plugins may be developed using the Cytoscape open Java software architecture by anyone and plugin community development is encouraged. Cytoscape also has a JavaScript-centric sister project named Cytoscape.js that can be used to analyse and visualise graphs in JavaScript environments, like a browser.
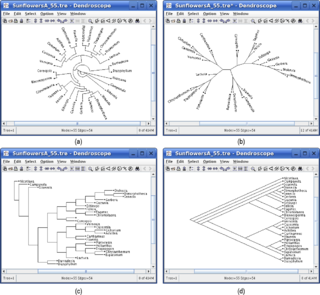 W
WDendroscope is an interactive computer software program written in Java for viewing Phylogenetic trees. This program is designed to view trees of all sizes and is very useful for creating figures. Dendroscope can be used for a variety of analyses of molecular data sets but is particularly designed for metagenomics or analyses of uncultured environmental samples.
DreamLab is a volunteer computing mobile Android and iOS app launched in 2015 by Imperial College London and the Vodafone Foundation.
 W
WGalaxy is a scientific workflow, data integration, and data and analysis persistence and publishing platform that aims to make computational biology accessible to research scientists that do not have computer programming or systems administration experience. Although it was initially developed for genomics research, it is largely domain agnostic and is now used as a general bioinformatics workflow management system.
 W
WGene Designer is a computer software package for bioinformatics. It is used by molecular biologists from academia, government, and the pharmaceutical, chemical, agricultural, and biotechnology industries to design, clone, and validate genetic sequences. It is proprietary software, released as freeware needing registration.
The Generic Model Organism Database (GMOD) project provides biological research communities with a toolkit of open-source software components for visualizing, annotating, managing, and storing biological data. The GMOD project is funded by the United States National Institutes of Health, National Science Foundation and the USDA Agricultural Research Service.
 W
WGenGIS merges geographic, ecological and phylogenetic biodiversity data in a single interactive visualization and analysis environment. A key feature of GenGIS is the testing of geographic axes that can correspond to routes of migration or gradients that influence community similarity. Data can also be explored using graphical summaries of data on a site-by-site basis, as 3D geophylogenies, or custom visualizations developed using a plugin framework. Standard statistical test such as linear regression and Mantel are provided, and the R statistical language can be accessed directly within GenGIS. Since its release, GenGIS has been used to investigate the phylogeography of viruses and bacteriophages, bacteria, and eukaryotes.
 W
WGenMAPP is a free, open-source bioinformatics software tool designed to visualize and analyze genomic data in the context of pathways, connecting gene-level datasets to biological processes and disease. First created in 2000, GenMAPP is developed by an open-source team based in an academic research laboratory. GenMAPP maintains databases of gene identifiers and collections of pathway maps in addition to visualization and analysis tools. Together with other public resources, GenMAPP aims to provide the research community with tools to gain insight into biology through the integration of data types ranging from genes to proteins to pathways to disease.
 W
WGENtle is a free software under GPL license.
 W
WHMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.
 W
WIMOD is an open-source, cross-platform suite of modeling, display and image processing programs used for 3D reconstruction and modeling of microscopy images with a special emphasis on electron microscopy data. IMOD has been used across a range of scales from macromolecule structures to organelles to whole cells and can also be used for optical sections. Included in IMOD are tools for image reconstruction, image segmentation, 3D mesh modeling and analysis of 2D and 3D data.
 W
WIntegrated Genome Browser (IGB) is an open-source genome browser, a visualization tool used to observe biologically-interesting patterns in genomic data sets, including sequence data, gene models, alignments, and data from DNA microarrays.
 W
WInterMine is an open source data warehouse system, licensed under the LGPL 2.1. InterMine is used to create databases of biological data accessed by sophisticated web query tools. InterMine can be used to create databases from a single data set or can integrate multiple sources of data. Support is provided for several common biological formats and there is a framework for adding other data. InterMine includes a user-friendly web interface that works 'out of the box' and can be easily customised.
The Kinetic Simulation Algorithm Ontology (KiSAO) supplies information about existing algorithms available for the simulation of systems biology models, their characterization and interrelationships. KiSAO is part of the BioModels.net project and of the COMBINE initiative.
 W
WIn bioinformatics, MAFFT is a program used to create multiple sequence alignments of amino acid or nucleotide sequences. Published in 2002, the first version of MAFFT used an algorithm based on progressive alignment, in which the sequences were clustered with the help of the Fast Fourier Transform. Subsequent versions of MAFFT have added other algorithms and modes of operation, including options for faster alignment of large numbers of sequences, higher accuracy alignments, alignment of non-coding RNA sequences, and the addition of new sequences to existing alignments.
 W
WMesquite is a software package primarily designed for phylogenetic analyses. It was developed as a successor to MacClade, when the authors recognized that implementing a modular architecture in MacClade would be infeasible. Mesquite is largely written in Java and uses NEXUS-formatted files as input. Mesquite is available as a compiled executable for Macintosh, Windows, and Unix-like platforms, and the source code is available on GitHub.
Molecular Evolutionary Genetics Analysis (MEGA) is computer software for conducting statistical analysis of molecular evolution and for constructing phylogenetic trees. It includes many sophisticated methods and tools for phylogenomics and phylomedicine. It is licensed as proprietary freeware. The project for developing this software was initiated by the leadership of Masatoshi Nei in his laboratory at the Pennsylvania State University in collaboration with his graduate student Sudhir Kumar and postdoctoral fellow Koichiro Tamura. Nei wrote a monograph (pp. 130) outlining the scope of the software and presenting new statistical methods that were included in MEGA. The entire set of computer programs was written by Kumar and Tamura. The personal computers then lacked the ability to send the monograph and software electronically, so they were delivered by postal mail. From the start, MEGA was intended to be easy-to-use and include solid statistical methods only.
OpenChrom is an open source software for the analysis and visualization of mass spectrometric and chromatographic data. Its focus is to handle native data files from several mass spectrometry systems, vendors like Agilent Technologies, Varian, Shimadzu, Thermo Fisher, PerkinElmer and others. But also data formats from other detector types are supported recently.
 W
WPSORT is a bioinformatics tool used for the prediction of protein localisation sites in cells. It receives the information of an amino acid sequence and its taxon of origin as inputs. Then it analyses the input sequence by applying the stored rules for various sequence features of known protein sorting signals. Finally, it reports the possibility for the input protein to be localised at each candidate site with additional information.
Rosetta@home is a distributed computing project for protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker laboratory at the University of Washington. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
Sequerome is a web-based sequence profiling tool for integrating the results of a BLAST sequence-alignment report with external research tools and servers that perform advanced sequence manipulations, and allowing the user to record the steps of such an analysis. Sequerome is a web-based Java tool that acts as a front-end to BLAST queries and provides simplified access to web-distributed resources for protein and nucleic acid analysis.
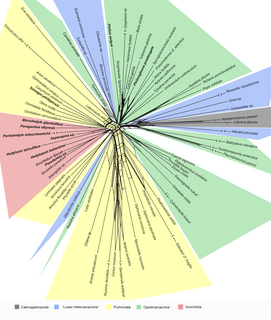 W
WSplitsTree is a popular free-ware program for inferring phylogenetic trees, phylogenetic networks, or, more generally, splits graphs, from various types of data such as a sequence alignment, a distance matrix or a set of trees. SplitsTree implements published methods such as split decomposition, neighbor-net, consensus networks, super networks methods or methods for computing hybridization or simple recombination networks. It uses the NEXUS file format, the splits graph is defined using a special data block.
TimeTree is a free public database developed by S. Blair Hedges and Sudhir Kumar, now at Temple University, for presenting times of divergence in the tree of life. The basic concept has been to produce and present a community consensus of the timetree of life from published studies, now numbering 3,998, and allow easy access to that information, on the web or mobile device. The database permits searching for average node times between two species or higher taxa and building of a timetree of life for a higher taxa. TimeTree has been used in public education to conceptualize the evolution of life, such as in high school settings. The original development of TimeTree dates to the late 1990s, with initial support from NASA Astrobiology Institute. Since then it has been supported by additional grants from NASA, and by NSF and NIH.
UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.
 W
WUTOPIA is a suite of free tools for visualising and analysing bioinformatics data. Based on an ontology-driven data model, it contains applications for viewing and aligning protein sequences, rendering complex molecular structures in 3D, and for finding and using resources such as web services and data objects. There are two major components, the protein analysis suite and UTOPIA documents.
 W
WXCMS Online is a cloud version of the original XCMS technology, created by the Siuzdak Lab at Scripps Research. XCMS introduced the concept of nonlinear retention time alignment that allowed for the statistical assessment of the detected peaks across LCMS and GCMS datasets. XCMS Online was designed to facilitate XCMS analyses through a cloud portal and as a more straightforward way to analyze, visualize and share untargeted metabolomic data. Further to this, the combination of XCMS and METLIN allows for the identification of known molecules using METLIN's tandem mass spectrometry data, and enables the identification of unknown via similarity searching of tandem mass spectrometry data. XCMS Online has also become a systems biology tool for integrating different omic data sets. XCMS Online and METLIN now host over 40,000 registered users.XCMSOnline