 W
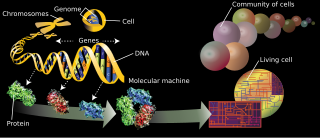
WGenomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
 W
WThe branches of science known informally as omics are various disciplines in biology whose names end in the suffix -omics, such as genomics, proteomics, metabolomics, and glycomics. Omics aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism or organisms.
 W
WThe three-base periodicity property in the field of Genomics is a property that is characteristic of protein-coding DNA sequences. The existence of this property can be shown by performing Fourier analysis on signals derived from segments of DNA sequences. Because of its predictive power, it has been used as a preliminary indicator in gene prediction.
 W
WThe $1,000 genome refers to an era of predictive and personalized medicine during which the cost of fully sequencing an individual's genome (WGS) is roughly one thousand USD. It is also the title of a book by British science writer and founding editor of Nature Genetics, Kevin Davies. By late 2015, the cost to generate a high-quality 'draft' whole human genome sequence was just below $1,500.
 W
WActivity-based proteomics, or activity-based protein profiling (ABPP) is a functional proteomic technology that uses chemical probes that react with mechanistically related classes of enzymes.
 W
WMaqsudul Alam was a Bangladeshi-born life-science scientist who is known for his work on genome sequencing. His work on genome sequencing started with bacteria Idiomarina loihiensis in 2003. He came into the focus of Bangladeshi people after his work on genome sequencing of tosha jute in 2010, white jute in 2013 and jute attacking fungus in 2012.
 W
WThe Allen Mouse and Human Brain Atlases are projects within the Allen Institute for Brain Science which seek to combine genomics with neuroanatomy by creating gene expression maps for the mouse and human brain. They were initiated in September 2003 with a $100 million donation from Paul G. Allen and the first atlas went public in September 2006. As of May 2012, seven brain atlases have been published: Mouse Brain Atlas, Human Brain Atlas, Developing Mouse Brain Atlas, Developing Human Brain Atlas, Mouse Connectivity Atlas, Non-Human Primate Atlas, and Mouse Spinal Cord Atlas. There are also three related projects with data banks: Glioblastoma, Mouse Diversity, and Sleep. It is the hope of the Allen Institute that their findings will help advance various fields of science, especially those surrounding the understanding of neurobiological diseases. The atlases are free and available for public use online.
The Archon Genomics X PRIZE presented by Express Scripts for Genomics, the second X Prize offered by the X Prize Foundation, based in Playa Vista, California, was announced on October 4, 2006 stating that the prize of "$10 million will be awarded to the first team to rapidly, accurately and economically sequence 100 whole human genomes to an unprecedented level of accuracy." The 30 day evaluation phase of the competition to begin on September 5, 2013, was canceled August 22, 2013 and this cancellation was debated on March 27, 2014.
 W
WIn molecular biology, biochips are essentially miniaturized laboratories that can perform hundreds or thousands of simultaneous biochemical reactions. Biochips enable researchers to quickly screen large numbers of biological analytes for a variety of purposes, from disease diagnosis to detection of bioterrorism agents. Digital microfluidic biochips have become one of the most promising technologies in many biomedical fields. In a digital microfluidic biochip, a group of (adjacent) cells in the microfluidic array can be configured to work as storage, functional operations, as well as for transporting fluid droplets dynamically.
 W
WBisulfite sequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
Blast2GO, first published in 2005, is a bioinformatics software tool for the automatic, high-throughput functional annotation of novel sequence data. It makes use of the BLAST algorithm to identify similar sequences to then transfers existing functional annotation from yet characterised sequences to the novel one. The functional information is represented via the Gene Ontology (GO), a controlled vocabulary of functional attributes. The Gene Ontology, or GO, is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
 W
WCG suppression is a term for the phenomenon that CG dinucleotides are very uncommon in most portions of vertebrate genomes.
 W
WChemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
 W
WChIP-exo is a chromatin immunoprecipitation based method for mapping the locations at which a protein of interest binds to the genome. It is a modification of the ChIP-seq protocol, improving the resolution of binding sites from hundreds of base pairs to almost one base pair. It employs the use of exonucleases to degrade strands of the protein-bound DNA in the 5'-3' direction to within a small number of nucleotides of the protein binding site. The nucleotides of the exonuclease-treated ends are determined using some combination of DNA sequencing, microarrays, and PCR. These sequences are then mapped to the genome to identify the locations on the genome at which the protein binds.
 W
WCofactor Genomics is a biotech company founded by past Human Genome Project Scientists. Cofactor's mission is to enable drug development, medical research, and personalized medicine using RNA-based information and technologies.
 W
WComparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
 W
WCopy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.
 W
WDelphibacteria is a candidate bacterial phylum in the FCB group. The phylum was first proposed after analysis of two genomes from the mouths of two bottlenose dolphins. "Dephibacteria" was proposed in recognition of the first genomic representatives having been recovered from the dolphin mouth. Members of the Delphibacteria phylum have been retroactively detected in a variety of environments.
 W
WThe double cut and join (DCJ) model is a model for genome rearrangement used to define an edit distance between genomes based on gene order and orientation, rather than nucleotide sequence. It takes the fundamental units of a genome to be synteny blocks, maximal sections of DNA conserved between genomes. It focuses on changes due to genome rearrangement operations such as inversions, translocations as well as the creation and absorption of circular intermediates.
 W
WDuplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double stranded DNA to detect mutations with higher accuracy and lower error rate. This method uses degenerate molecular tags in addition to sequencing adapters to recognize reads originating from each strand of DNA. The generated sequencing reads then will be analyzed using two methods: single strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly. Duplex sequencing theoretically can detect mutations with frequencies as low as 5 x 10−8 that is more than 10,000 fold higher in accuracy compared to the conventional next-generation sequencing methods.
 W
WFunctional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.
The Generic Model Organism Database (GMOD) project provides biological research communities with a toolkit of open-source software components for visualizing, annotating, managing, and storing biological data. The GMOD project is funded by the United States National Institutes of Health, National Science Foundation and the USDA Agricultural Research Service.
 W
WIn the fields of molecular biology and genetics, a genome is all genetic material of an organism. It consists of DNA. The genome includes both the genes and the noncoding DNA, as well as mitochondrial DNA and chloroplast DNA. The study of the genome is called genomics.
 W
WGenome evolution is the process by which a genome changes in structure (sequence) or size over time. The study of genome evolution involves multiple fields such as structural analysis of the genome, the study of genomic parasites, gene and ancient genome duplications, polyploidy, and comparative genomics. Genome evolution is a constantly changing and evolving field due to the steadily growing number of sequenced genomes, both prokaryotic and eukaryotic, available to the scientific community and the public at large.
 W
WGenome size is the total amount of DNA contained within one copy of a single complete genome. It is typically measured in terms of mass in picograms or less frequently in daltons, or as the total number of nucleotide base pair ed Mb or Mbp). One picogram is equal to 978 megabases. In diploid organisms, genome size is often used interchangeably with the term C-value.
 W
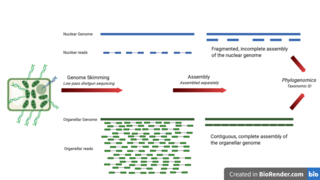
WGenome skimming is a sequencing approach that uses low-pass, shallow sequencing of a genome, to generate fragments of DNA, known as genome skims. These genome skims contain information about the high-copy fraction of the genome. The high-copy fraction of the genome consists of the ribosomal DNA, plastid genome (plastome), mitochondrial genome (mitogenome), and nuclear repeats such as microsatellites and transposable elements. It employs high-throughput, next generation sequencing technology to generate these skims. Although these skims are merely 'the tip of the genomic iceberg', phylogenomic analysis of them can still provide insights on evolutionary history and biodiversity at a lower cost and larger scale than traditional methods. Due to the small amount of DNA required for genome skimming, its methodology can be applied in other fields other than genomics. Tasks like this include determining the traceability of products in the food industry, enforcing international regulations regarding biodiversity and biological resources, and forensics.
 W
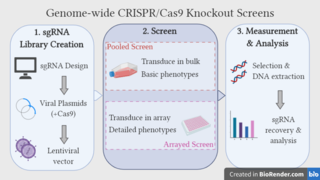
WGenome-wide CRISPR-Cas9 knockout screens aim to elucidate the relationship between genotype and phenotype by ablating gene expression on a genome-wide scale and studying the resulting phenotypic alterations. The approach utilises the CRISPR-Cas9 gene editing system, coupled with libraries of single guide RNAs (sgRNAs), which are designed to target every gene in the genome. Over recent years, the genome-wide CRISPR screen has emerged as a powerful tool for performing large-scale loss-of-function screens, with low noise, high knockout efficiency and minimal off-target effects.
 W
WBirds are the group of amniotes with the smallest genomes. Whereas mammal and reptilian genomes range between 1.0 and 8.2 giga base pairs (Gb), bird genomes have sizes between 0.91 Gb and 1.3 Gb. Just as happens to any other living being, bird genomes’ reflect the action of natural selection upon these animals. Their genomes are the basis of their morphology and behaviour.
 W
WThe hereditary material i.e. DNA of an organism is composed of a sequence of four nucleotides in a specific pattern, which encode information as a function of their order. Genomic organization refers to the linear order of DNA elements and their division into chromosomes. "Genome organization" can also refer to the 3D structure of chromosomes and the positioning of DNA sequences within the nucleus.
 W
WThe human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome, and the mitochondrial genome. Human genomes include both protein-coding DNA genes and noncoding DNA. Haploid human genomes, which are contained in germ cells consist of three billion DNA base pairs, while diploid genomes have twice the DNA content. While there are significant differences among the genomes of human individuals, these are considerably smaller than the differences between humans and their closest living relatives, the bonobos and chimpanzees.
 W
WCSIR Institute of Genomics and Integrative Biology (CSIR-IGIB) is a scientific research institute devoted primarily to biological research. It is a part of Council of Scientific and Industrial Research (CSIR), India.
 W
WThe International Grape Genomics Program (IGGP) is a collaborative genome project dedicated to determining the genome sequence of the grapevine Vitis vinifera. It is a multinational project involving research centers in Australia, Canada, Chile, France, Germany, Italy, South Africa, Spain, and the United States.
 W
WGerardo Jiménez Sánchez is a Mexican-born pediatrician, scientist and businessman. Along with David Valle and Barton Childs, he completed the first medical analysis of the human genome. He was founder and director of the first National Institute of Genomic Medicine in Latin America (INMEGEN) and leader of the team that developed the Genomic Map of the Mexican Population.
 W
WLinear Amplification via Transposon Insertion (LIANTI) is a linear whole genome amplification (WGA) method. To analyze or sequence very small amount of DNA, i.e. genomic DNA from a single cell, the picograms of DNA is subject to WGA to amplify at least thousands of times into nanogram scale, before DNA analysis or sequencing can be carried out. Previous WGA methods use exponential/nonlinear amplification schemes, leading to bias accumulation and error propagation. LIANTI achieved linear amplification of the whole genome for the first time, enabling more uniform and accurate amplification.
 W
WMedicago truncatula, the barrelclover, strong-spined medick, barrel medic, or barrel medick, is a small annual legume native to the Mediterranean region that is used in genomic research. It is a low-growing, clover-like plant 10–60 centimetres (3.9–23.6 in) tall with trifoliate leaves. Each leaflet is rounded, 1–2 centimetres (0.39–0.79 in) long, often with a dark spot in the center. The flowers are yellow, produced singly or in a small inflorescence of two to five together; the fruit is a small, spiny pod.
 W
WMetabolic network reconstruction and simulation allows for an in-depth insight into the molecular mechanisms of a particular organism. In particular, these models correlate the genome with molecular physiology. A reconstruction breaks down metabolic pathways into their respective reactions and enzymes, and analyzes them within the perspective of the entire network. In simplified terms, a reconstruction collects all of the relevant metabolic information of an organism and compiles it in a mathematical model. Validation and analysis of reconstructions can allow identification of key features of metabolism such as growth yield, resource distribution, network robustness, and gene essentiality. This knowledge can then be applied to create novel biotechnology.
 W
WMetagenomics is the study of genetic material recovered directly from environmental samples. The broad field may also be referred to as environmental genomics, ecogenomics or community genomics.
 W
WKaren Elizabeth Hayden Miga is an American genomics expert who leads the Telomere-to-Telomore (T2T) consortium that seeks to fully complete the assembly of the human genome. She serves as an Assistant Research Scientist at the University of California, Santa Cruz. She was named as "One to Watch" in the 2020 Nature's 10.
 W
WThe Min System is a mechanism composed of three proteins MinC, MinD, and MinE used by E. coli as a means of properly localizing the septum prior to cell division. Each component participates in generating a dynamic oscillation of FtsZ protein inhibition between the two bacterial poles to precisely specify the mid-zone of the cell, allowing the cell to accurately divide in two. This system is known to function in conjunction with a second negative regulatory system, the nucleoid occlusion system (NO), to ensure proper spatial and temporal regulation of chromosomal segregation and division.
 W
WMolecular models of DNA structures are representations of the molecular geometry and topology of deoxyribonucleic acid (DNA) molecules using one of several means, with the aim of simplifying and presenting the essential, physical and chemical, properties of DNA molecular structures either in vivo or in vitro. These representations include closely packed spheres made of plastic, metal wires for skeletal models, graphic computations and animations by computers, artistic rendering. Computer molecular models also allow animations and molecular dynamics simulations that are very important for understanding how DNA functions in vivo.
 W
WThe Neuroimaging Tools and Resources Collaboratory is a neuroimaging informatics knowledge environment for MR, PET/SPECT, CT, EEG/MEG, optical imaging, clinical neuroinformatics, imaging genomics, and computational neuroscience tools and resources.
 W
WThe Personal Genetics Education Project (pgEd) aims to engage and inform a worldwide audience about the benefits of knowing one's genome as well as the ethical, legal and social issues (ELSI) and dimensions of personal genetics. pgEd was founded in 2006, is housed in the Department of Genetics at Harvard Medical School and is directed by Ting Wu, a professor in that department. It employs a variety of strategies for reaching general audiences, including generating online curricular materials, leading discussions in classrooms, workshops, and conferences, developing a mobile educational game (Map-Ed), holding an annual conference geared toward accelerating awareness (GETed), and working with the world of entertainment to improve accuracy and outreach.
WPharmacogenomics is the study of the role of the genome in drug response. Its name reflects its combining of pharmacology and genomics. Pharmacogenomics analyzes how the genetic makeup of an individual affects his/her response to drugs. It deals with the influence of acquired and inherited genetic variation on drug response in patients by correlating gene expression or single-nucleotide polymorphisms with pharmacokinetics and pharmacodynamics. The term pharmacogenomics is often used interchangeably with pharmacogenetics. Although both terms relate to drug response based on genetic influences, pharmacogenetics focuses on single drug-gene interactions, while pharmacogenomics encompasses a more genome-wide association approach, incorporating genomics and epigenetics while dealing with the effects of multiple genes on drug response.
 W
WThe Pathogen-Host Interaction database (PHI-base) is a biological database that contains curated information on genes experimentally proven to affect the outcome of pathogen-host interactions. The database is maintained by researchers at Rothamsted Research, together with external collaborators since 2005. Since April 2017 PHI-base is part of ELIXIR, the European life-science infrastructure for biological information via its ELIXIR-UK node.
 W
WProteogenomics is a field of biological research that utilizes a combination of proteomics, genomics, and transcriptomics to aid in the discovery and identification of peptides. Proteogenomics is used to identify new peptides by comparing MS/MS spectra against a protein database that has been derived from genomic and transcriptomic information. Proteogenomics often refers to studies that use proteomic information, often derived from mass spectrometry, to improve gene annotations. Genomics deals with the genetic code of entire organisms, while transcriptomics deals with the study of RNA sequencing and transcripts. Proteomics utilizes tandem mass spectrometry and liquid chromatography to identify and study the functions of proteins. Proteomics is being utilized to discover all the proteins expressed within an organism, known as its proteome. The issue with proteomics is that it relies on the assumption that current gene models are correct and that the correct protein sequences can be found using a reference protein sequence database; however, this is not always the case as some peptides cannot be located in the database. In addition, novel protein sequences can occur through mutations. these issues can be fixed with the use of proteomic, genomic, and trancriptomic data. The utilization of both proteomics and genomics led to proteogenomics which became its own field in 2004..
 W
WP3G (Public Population Project in Genomics and Society) is a not-for-profit international consortium dedicated to facilitating collaboration between researchers and biobanks working in the area of human population genomics. P3G is member-based and composed of experts from the different disciplines in the areas of and related to genomics, including epidemiology, law, ethics, technology, biomolecular science, etc. P3G and its members are committed to a philosophy of information sharing with the goal of supporting researchers working in areas that will improve the health of people around the world.
 W
WReduced representation bisulfite sequencing (RRBS) is an efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level. It combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content. Due to the high cost and depth of sequencing to analyze methylation status in the entire genome, Meissner et al. developed this technique in 2005 to reduce the amount of nucleotides required to sequence to 1% of the genome. The fragments that comprise the reduced genome still include the majority of promoters, as well as regions such as repeated sequences that are difficult to profile using conventional bisulfite sequencing approaches.
 W
WSingle cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.
 W
WPost-genomic research has rendered bacterial small non-coding RNAs (sRNAs) as major players in post-transcriptional regulation of gene expression in response to environmental stimuli. The α-subdivision of the Proteobacteria includes Gram-negative microorganisms with diverse life styles; frequently involving long-term interactions with higher eukaryotes.
 W
WStructural genomics seeks to describe the 3-dimensional structure of every protein encoded by a given genome. This genome-based approach allows for a high-throughput method of structure determination by a combination of experimental and modeling approaches. The principal difference between structural genomics and traditional structural prediction is that structural genomics attempts to determine the structure of every protein encoded by the genome, rather than focusing on one particular protein. With full-genome sequences available, structure prediction can be done more quickly through a combination of experimental and modeling approaches, especially because the availability of large number of sequenced genomes and previously solved protein structures allows scientists to model protein structure on the structures of previously solved homologs.
 W
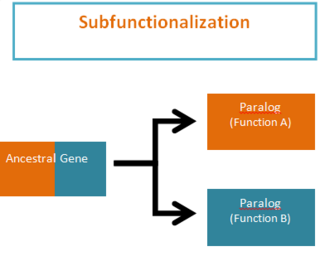
WSubfunctionalization was proposed by Stoltzfus (1999) and Force et al. (1999) as one of the possible outcomes of functional divergence that occurs after a gene duplication event, in which pairs of genes that originate from duplication, or paralogs, take on separate functions. Subfunctionalization is a neutral mutation process; meaning that no new adaptations are formed. During the process of gene duplication paralogs simply undergo a division of labor by retaining different parts (subfunctions) of their original ancestral function. This partitioning event occurs because of segmental gene silencing leading to the formation of paralogs that are no longer duplicates, because each gene only retains a single function. It is important to note that the ancestral gene was capable of performing both functions and the descendant duplicate genes can now only perform one of the original ancestral functions.
 W
WTakifugu rubripes, commonly known as the Japanese puffer, Tiger puffer, or torafugu, is a pufferfish in the genus Takifugu. It is distinguished by a very small genome that has been fully sequenced because of its use as a model species and is in widespread use as a reference in genomics.
 W
WThe thanatotranscriptome denotes all RNA from the transcript of the part of genome still active or awakened in the internal organs of a dead body for 24 to 48 hours following the time of the death. ·
WViral metagenomics is the study of viral genetic material sourced directly from the environment rather than from a host or natural reservoir. The goal is to ascertain the viral diversity in the environment that is often missed in studies targeting specific potential reservoirs. It reveals important information on virus evolution and the genetic diversity of the viral community without the need for isolating viral species and cultivating them in the laboratory. With the new techniques available that exploit next-generation sequencing (NGS), it is possible to study the virome of some ecosystems, even if the analysis still has some issues, in particular the lack of universal markers. Some of the first metagenomic studies of viruses were done with ocean samples, and revealed that most of the sequences of DNA and RNA viruses had no matches in databases. Subsequently, some studies about the soil virome were performed with a particular interest on bacteriophages, and it was discovered that there are almost the same number of viruses and bacteria. This approach has created improvements in molecular epidemiology and accelerated the discovery of novel viruses.
 W
WWhole genome sequencing is ostensibly the process of determining the complete DNA sequence of an organism's genome at a single time. This entails sequencing all of an organism's chromosomal DNA as well as DNA contained in the mitochondria and, for plants, in the chloroplast. In practice, genome sequences that are nearly complete are also called whole genome sequences.
 W
W