 W
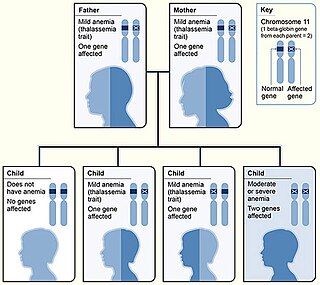
WAlpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.
 W
WBeta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.
 W
WDelta-beta thalassemia is a form of thalassemia, and is autosomal recessive in terms of heredity. It is associated with hemoglobin subunit delta.
 W
WGlucose-6-phosphate dehydrogenase deficiency (G6PDD) is an inborn error of metabolism that predisposes to red blood cell breakdown. Most of the time, those who are affected have no symptoms. Following a specific trigger, symptoms such as yellowish skin, dark urine, shortness of breath, and feeling tired may develop. Complications can include anemia and newborn jaundice. Some people never have symptoms.
 W
WHereditary elliptocytosis, also known as ovalocytosis, is an inherited blood disorder in which an abnormally large number of the person's red blood cells are elliptical rather than the typical biconcave disc shape. Such morphologically distinctive erythrocytes are sometimes referred to as elliptocytes or ovalocytes. It is one of many red-cell membrane defects. In its severe forms, this disorder predisposes to haemolytic anaemia. Although pathological in humans, elliptocytosis is normal in camelids.
 W
WHereditary spherocytosis is an abnormality of red blood cells, or erythrocytes. The disorder is caused by mutations in genes relating to membrane proteins that allow for the erythrocytes to change shape. The abnormal erythrocytes are sphere-shaped (spherocytosis) rather than the normal biconcave disk shaped. Dysfunctional membrane proteins interfere with the cell's ability to be flexible to travel from the arteries to the smaller capillaries. This difference in shape also makes the red blood cells more prone to rupture. Cells with these dysfunctional proteins are degraded in the spleen. This shortage of erythrocytes results in hemolytic anemia.
 W
WHereditary stomatocytosis describes a number of inherited autosomal dominant human conditions which affect the red blood cell, in which the membrane or outer coating of the cell 'leaks' sodium and potassium ions.
 W
WPyruvate kinase deficiency is an inherited metabolic disorder of the enzyme pyruvate kinase which affects the survival of red blood cells. Both autosomal dominant and recessive inheritance have been observed with the disorder; classically, and more commonly, the inheritance is autosomal recessive. Pyruvate kinase deficiency is the second most common cause of enzyme-deficient hemolytic anemia, following G6PD deficiency.
 W
WSickle cell trait describes a condition in which a person has one abnormal allele of the hemoglobin beta gene, but does not display the severe symptoms of sickle cell disease that occur in a person who has two copies of that allele. Those who are heterozygous for the sickle cell allele produce both normal and abnormal hemoglobin.
 W
WSickle cell disease (SCD) is a group of blood disorders typically inherited from a person's parents. The most common type is known as sickle cell anaemia (SCA). It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells. This leads to a rigid, sickle-like shape under certain circumstances. Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain, anemia, swelling in the hands and feet, bacterial infections and stroke. Long-term pain may develop as people get older. The average life expectancy in the developed world is 40 to 60 years.
WThalassemias are inherited blood disorders characterized by decreased hemoglobin production. Symptoms depend on the type and can vary from none to severe. Often there is mild to severe anemia. Anemia can result in feeling tired and pale skin. There may also be bone problems, an enlarged spleen, yellowish skin, and dark urine. Slow growth may occur in children.
 W
WTriosephosphate isomerase deficiency is a rare autosomal recessive metabolic disorder which was initially described in 1965.