 W
WAndrogen insensitivity syndrome (AIS) is an intersex condition occurring in 1:20,000 individuals to 1:64,000, resulting in the partial or complete inability of the cell to respond to androgens. The unresponsiveness of the cell to the presence of androgenic hormones can impair or prevent the masculinization of male genitalia in the developing fetus, as well as impairing or preventing the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development. As such, the insensitivity to androgens is clinically significant only when it occurs in genetic males. Clinical phenotypes in these individuals range from a typical male habitus with mild spermatogenic defect or reduced secondary terminal hair, to a full female habitus, despite the presence of a Y-chromosome.
 W
WAromatase excess syndrome is a rare genetic and endocrine syndrome which is characterized by an overexpression of aromatase, the enzyme responsible for the biosynthesis of the estrogen sex hormones from the androgens, in turn resulting in excessive levels of circulating estrogens and, accordingly, symptoms of hyperestrogenism. It affects both sexes, manifesting itself in males as marked or complete phenotypical feminization and in females as hyperfeminization.
 W
WBarakat syndrome, is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It was first described by Amin J. Barakat et al. in 1977.
 W
WCushing's syndrome is the collection of signs and symptoms due to prolonged exposure to glucocorticoids such as cortisol. Signs and symptoms may include high blood pressure, abdominal obesity but with thin arms and legs, reddish stretch marks, a round red face, a fat lump between the shoulders, weak muscles, weak bones, acne, and fragile skin that heals poorly. Women may have more hair and irregular menstruation. Occasionally there may be changes in mood, headaches, and a chronic feeling of tiredness.
 W
WEmpty sella syndrome is the condition when the pituitary gland shrinks or becomes flattened, filling the sella turcica with cerebrospinal fluid instead of the normal pituitary. It can be discovered as part of the diagnostic workup of pituitary disorders, or as an incidental finding when imaging the brain.
 W
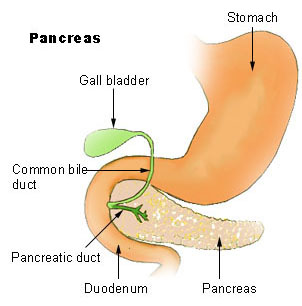
WJohanson–Blizzard syndrome is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with mental retardation, hearing loss and growth failure. It is sometimes described as a form of ectodermal dysplasia.
 W
WKowarski syndrome describes cases of growth failure, despite the presence of normal or slightly high blood growth hormone by radioimmunoassay (RIA-GH) and low serum IGF1, and who exhibit a significant increase in growth rate following recombinant GH therapy.
 W
WMorgagni-Stewart-Morel syndrome is a condition with a wide range of associated endocrine problems including: diabetes mellitus, diabetes insipidus, and hyperparathyroidism. Other signs and symptoms include headaches, vertigo, hirsutism, menstrual disorder, galactorrhoea, obesity, depression, and seizures. Thickening of the inner table of the frontal part of the skull a usually benign condition known as hyperostosis frontalis interna. The syndrome was first described in 1765. It is named after the Italian anatomist and pathologist Giovanni Battista Morgagni, the British neurologist Roy Mackenzie Stewart, and the Swiss psychiatrist Ferdinand Morel.
 W
WMultiple endocrine neoplasia type 2 is a group of medical disorders associated with tumors of the endocrine system. The tumors may be benign or malignant (cancer). They generally occur in endocrine organs, but may also occur in endocrine tissues of organs not classically thought of as endocrine.
 W
WPituitary stalk interruption syndrome (PSIS) is a congenital disorder characterised by the triad of an absent or exceedingly thin pituitary stalk, an ectopic or absent posterior pituitary and/or absent or hypoplastic anterior pituitary.
 W
WPrimary pigmented nodular adrenocortical disease (PPNAD) was first coined in 1984 by Carney et al. it often occurs in association with Carney complex (CNC). CNC is a rare syndrome that involves the formation of abnormal tumours that cause endocrine hyperactivity.
 W
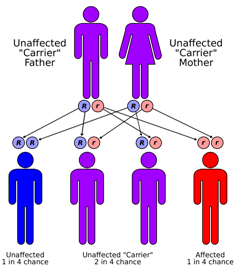
WSanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
 W
WTriple-A syndrome or AAA syndrome, is a rare autosomal recessive congenital disorder. In most cases, there is no family history of it. The syndrome was first identified by Jeremy Allgrove and colleagues in 1978, since then just over 100 cases have been reported. The syndrome involves achalasia, addisonianism, and alacrima. Alacrima is usually the earliest manifestation. It is a progressive disorder that can take years to develop the full-blown clinical picture.