 W
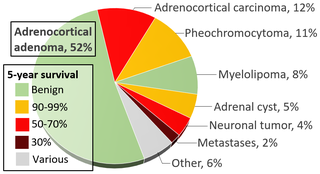
WAn adrenal tumor or adrenal mass is any benign or malignant neoplasms of the adrenal gland, several of which are notable for their tendency to overproduce endocrine hormones. Adrenal cancer is the presence of malignant adrenal tumors, and includes neuroblastoma, adrenocortical carcinoma and some adrenal pheochromocytomas. Most adrenal pheochromocytomas and all adrenocortical adenomas are benign tumors, which do not metastasize or invade nearby tissues, but may cause significant health problems by unbalancing hormones.
 W
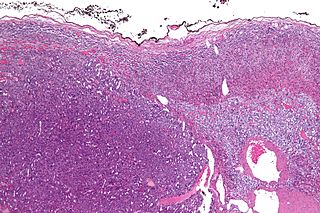
WAdrenocortical adenoma is commonly described as a benign neoplasm emerging from the cells that comprise the adrenal cortex. Like most adenomas, the adrenocortical adenoma is considered a benign tumor since the majority of them are non-functioning and asymptomatic. Adrenocortical adenomas are classified as ACTH-independent disorders, and are commonly associated with conditions linked to hyperadrenalism such as Cushing's syndrome (hypercortisolism) or Conn's syndrome (hyperaldosteronism), which is also known as primary aldosteronism. In addition, recent case reports further support the affiliation of adrenocortical adenomas with hyperandrogenism or florid hyperandrogenism which can cause hyperandrogenic hirsutism in females. "Cushing's syndrome" differs from the "Cushing's disease" even though both conditions are induced by hypercortisolism. The term "Cushing's disease" refers specifically to "secondary hypercortisolism" classified as "ACTH-dependent Cushing's syndrome" caused by pituitary adenomas. In contrast, "Cushing's syndrome" refers specifically to "primary hypercortisolism" classified as "ACTH-independent Cushing's syndrome" caused by adrenal adenomas.
 W
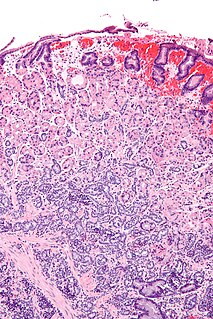
WAdrenocortical carcinoma (ACC) is an aggressive cancer originating in the cortex of the adrenal gland.
 W
WA gastrinoma is a tumor derived from G cells in the duodenum, pancreas or less commonly stomach, that secretes the peptide hormone gastrin. There is hypersecretion of HCl acid into the duodenum, which causes the ulcers. Excessive HCl acid production also causes hyperperistalsis, and inhibits the activity of lipase, causing severe diarrhea.
 W
WAn insulinoma is a tumor of the pancreas that is derived from beta cells and secretes insulin. It is a rare form of a neuroendocrine tumor. Most insulinomas are benign in that they grow exclusively at their origin within the pancreas, but a minority metastasize. Insulinomas are one of the functional pancreatic neuroendocrine tumor (PNET) group. In the Medical Subject Headings classification, insulinoma is the only subtype of "islet cell adenoma".
 W
WLeydig cell tumour, also Leydig cell tumor, (testicular) interstitial cell tumour and (testicular) interstitial cell tumor, is a member of the sex cord-stromal tumour group of ovarian and testicular cancers. It arises from Leydig cells. While the tumour can occur at any age, it occurs most often in young adults.
 W
WThe term multiple endocrine neoplasia encompasses several distinct syndromes featuring tumors of endocrine glands, each with its own characteristic pattern. In some cases, the tumors are malignant, in others, benign. Benign or malignant tumors of nonendocrine tissues occur as components of some of these tumor syndromes.
 W
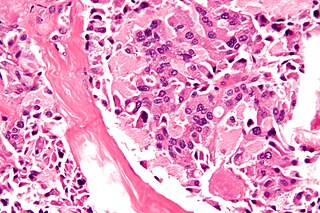
WMultiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. It was first described by Paul Wermer in 1954.
 W
WMultiple endocrine neoplasia type 2B is a genetic disease that causes multiple tumors on the mouth, eyes, and endocrine glands. It is the most severe type of multiple endocrine neoplasia, differentiated by the presence of benign oral and submucosal tumors in addition to endocrine malignancies. It was first described by Wagenmann in 1922, and was first recognized as a syndrome in 1965-1966 by E.D. Williams and D.J. Pollock.
 W
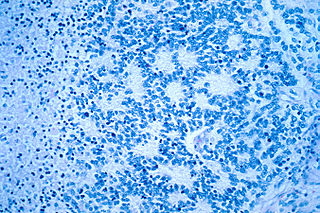
WNeuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue. It most frequently starts from one of the adrenal glands but can also develop in the neck, chest, abdomen, or spine. Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.
 W
WNeuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung and the rest of the body.
 W
WPapillary tumors of the pineal region (PTPR) were first described by A. Jouvet et al. in 2003 and were introduced in the World Health Organization (WHO) classification of Central Nervous System (CNS) in 2007. Papillary Tumors of the Pineal Region are located on the pineal gland which is located in the center of the brain. The pineal gland is located on roof of the diencephalon. It is a cone shaped structure dorsal to the midbrain tectum. The tumor appears to be derived from the specialized ependymal cells of the subcommissural organ. Papillary tumors of the central nervous system and particularly of the pineal region are very rare and so diagnosing them is extremely difficult.
 W
WA paraganglioma is a rare neuroendocrine neoplasm that may develop at various body sites. When the same type of tumor is found in the adrenal gland, they are referred to as a pheochromocytoma. They are rare tumors, with an overall estimated incidence of 1/300,000. Unlike other types of cancer, there is no test that determines benign from malignant tumors; long-term followup is therefore recommended for all individuals with paraganglioma.
 W
WA parathyroid adenoma is a benign tumor of the parathyroid gland. It generally causes hyperparathyroidism; there are very few reports of parathyroid adenomas that were not associated with hyperparathyroidism.
 W
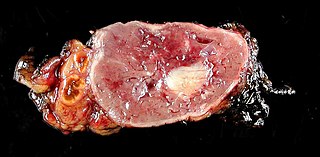
WParathyroid carcinoma is a rare cancer resulting in parathyroid adenoma to carcinoma progression. It forms in tissues of one or more of the parathyroid glands.
 W
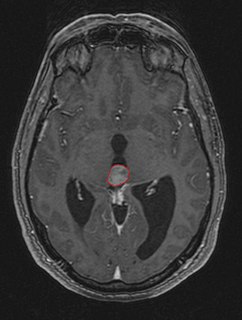
WPheochromocytoma is a rare, chromaffin cell tumor of the adrenal medulla. When a tumor composed of the same cells as a pheochromocytoma develops outside the adrenal gland, it is referred to as a paraganglioma. These neuroendocrine tumors are capable of producing and releasing massive amounts of catecholamines, metanephrines, or methoxytyramine, which result in the most common symptoms, including hypertension, tachycardia, and diaphoresis (sweating). However, not all of these tumors will secrete catecholamines. Those that do not are referred to as biochemically silent, and are predominantly located in the head and neck. While patients with biochemically silent disease will not suffer from the typical disease manifestations described above, the tumors grow and compress the surrounding structures of the head and neck, and can result in pulsatile tinnitus, hearing loss, aural fullness, dyspnea, and hoarseness. While tumors of the head and neck are parasympathetic, their sympathetic counterparts are predominantly located in the abdomen and pelvis, particularly concentrated at the organ of Zuckerkandl.
 W
WA pinealoma is a tumor of the pineal gland, a part of the brain that produces melatonin. If a pinealoma destroys the cells of the pineal gland in a child, it can cause precocious puberty.
 W
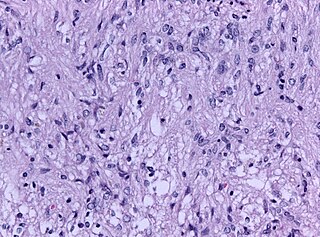
WPineocytoma, is a benign, slowly growing tumor of the pineal gland. Unlike the similar condition pineal gland cyst, it is uncommon.
 W
WPituicytoma is a rare brain tumor. It grows at the base of the brain from the pituitary gland. This tumor is thought to be derived from the parenchymal cells of the posterior lobe of the pituitary gland, called pituicytes. Some researchers believe that they arise from the folliculostellate cells in the anterior lobe of the pituitary. As such, it is a low-grade glioma. It occurs in adults and symptoms include visual disturbance and endocrine dysfunction. They are often mistaken for pituitary adenomas which have a similar presentation and occur in the same location. The treatment consists of surgical resection, which is curative in most cases.
 W
WPituitary adenomas are tumors that occur in the pituitary gland. Pituitary adenomas are generally divided into three categories dependent upon their biological functioning: benign adenoma, invasive adenoma, and carcinomas. Most adenomas are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms and the estimated prevalence rate in the general population is approximately 17%.
 W
WA prolactinoma is a benign tumor (adenoma) of the pituitary gland that produces a hormone called prolactin. It is the most common type of functioning pituitary tumor. Symptoms of prolactinoma are due to too much prolactin in the blood (hyperprolactinemia), or those caused by pressure of the tumor on surrounding tissues.
 W
WThyroid neoplasm is a neoplasm or tumor of the thyroid. It can be a benign tumor such as thyroid adenoma, or it can be a malignant neoplasm, such as papillary, follicular, medullary or anaplastic thyroid cancer. Most patients are 25 to 65 years of age when first diagnosed; women are more affected than men. The estimated number of new cases of thyroid cancer in the United States in 2010 is 44,670 compared to only 1,690 deaths. Of all thyroid nodules discovered, only about 5 percent are cancerous, and under 3 percent of those result in fatalities.