 W
W5α-Reductase deficiency is an autosomal recessive intersex condition caused by a mutation in SRD5A2, a gene encoding the enzyme 5α-reductase type 2. The phenotype this usually causes is pseudovaginal perineoscrotal hypospadias, a configuration of the external genitalia of an infant. In a sense, this configuration is roughly midway between phenotypical human male genitalia, and phenotypical human female genitalia, in structure and appearance. It is a relatively common form of genital ambiguity caused by undervirilization of genetic males due to several different intersex conditions.
 W
W17β-Hydroxysteroid dehydrogenase III deficiency is a rare autosomal recessive disorder of sexual development, or intersex condition, affecting testosterone biosynthesis by 17β-hydroxysteroid dehydrogenase III, which can produce impaired virilization of genetically male infants.
 W
WAndrogen insensitivity syndrome (AIS) is an intersex condition occurring in 1:20,000 individuals to 1:64,000, resulting in the partial or complete inability of the cell to respond to androgens. The unresponsiveness of the cell to the presence of androgenic hormones can impair or prevent the masculinization of male genitalia in the developing fetus, as well as impairing or preventing the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development. As such, the insensitivity to androgens is clinically significant only when it occurs in genetic males. Clinical phenotypes in these individuals range from a typical male habitus with mild spermatogenic defect or reduced secondary terminal hair, to a full female habitus, despite the presence of a Y-chromosome.
WComplete androgen insensitivity syndrome (CAIS) is a condition that results in the complete inability of the cell to respond to androgens. As such, the insensitivity to androgens is only clinically significant when it occurs in individuals with a Y chromosome or, more specifically, an SRY gene. The unresponsiveness of the cell to the presence of androgenic hormones prevents the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does allow, without significant impairment, female genital and sexual development in those with the condition.
WMild androgen insensitivity syndrome (MAIS) is a condition that results in a mild impairment of the cell's ability to respond to androgens. The degree of impairment is sufficient to impair spermatogenesis and / or the development of secondary sexual characteristics at puberty in males, but does not affect genital differentiation or development. Female genital and sexual development is not significantly affected by the insensitivity to androgens; as such, MAIS is only diagnosed in males. The clinical phenotype associated with MAIS is a normal male habitus with mild spermatogenic defect and / or reduced secondary terminal hair.
WPartial androgen insensitivity syndrome (PAIS) is a condition that results in the partial inability of the cell to respond to androgens. It is an X linked recessive condition. The partial unresponsiveness of the cell to the presence of androgenic hormones impairs the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development. As such, the insensitivity to androgens is clinically significant only when it occurs in individuals with a Y chromosome. Clinical features include ambiguous genitalia at birth and primary amenhorrhoea with clitoromegaly with inguinal masses. Mullerian structures are not present in the individual.
 W
WAromatase deficiency is a very rare condition characterised by the extremely low or absence of the enzyme aromatase activity in the body. It is an autosomal recessive disease resulted from various mutations of gene CPY19 (P450arom) which can lead to delayed puberty in females, osteoporosis in males and virilisation in pregnant mothers. As of 2016, only 35 cases have been described in medical literature.
WAromatase excess syndrome is a rare genetic and endocrine syndrome which is characterized by an overexpression of aromatase, the enzyme responsible for the biosynthesis of the estrogen sex hormones from the androgens, in turn resulting in excessive levels of circulating estrogens and, accordingly, symptoms of hyperestrogenism. It affects both sexes, manifesting itself in males as marked or complete phenotypical feminization and in females as hyperfeminization.
 W
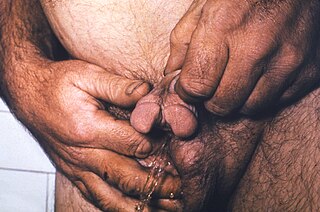
WA bifid penis is a rare congenital defect where two genital tubercles develop.
 W
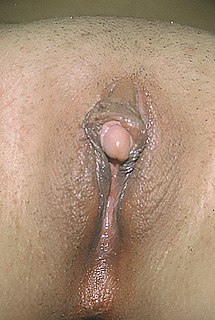
WClitoromegaly is an abnormal enlargement of the clitoris that is mostly congenital or acquired, though deliberately induced clitoris enlargement as a form of female genital body modification is achieved through various uses of anabolic steroids, including testosterone, and may also be referred to as clitoromegaly. Clitoromegaly is not the same as normal enlargement of the clitoris seen during sexual arousal.
 W
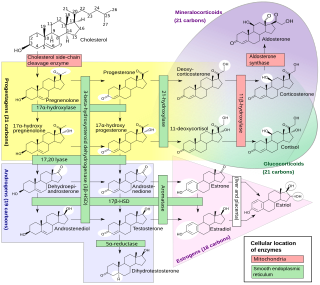
WCongenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene for one of the key enzymes in cortisol synthesis by the adrenal gland, 3β-hydroxysteroid dehydrogenase (3β-HSD) type II (HSD3B2). As a result, higher levels of 17α-hydroxypregnenolone appear in the blood with adrenocorticotropic hormone (ACTH) challenge, which stimulates adrenal corticosteroid synthesis.
 W
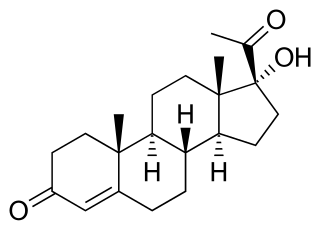
WCongenital adrenal hyperplasia due to 11β-hydroxylase deficiency is a form of congenital adrenal hyperplasia (CAH) which produces a higher than normal amount of androgen, resulting from a defect in the gene encoding the enzyme steroid 11β-hydroxylase (11β-OH) which mediates the final step of cortisol synthesis in the adrenal. 11β-OH CAH results in hypertension due to excessive mineralocorticoid effects. It also causes excessive androgen production both before and after birth and can virilize a genetically female fetus or a child of either sex.
 W
WCongenital adrenal hyperplasia due to 21-hydroxylase deficiency in all its forms, accounts for over 95% of diagnosed cases of congenital adrenal hyperplasia (CAH), and CAH in most contexts refers to 21-hydroxylase deficiency and different mutations related to enzyme impairment have been mapped on protein structure.
 W
WDiphallia, penile duplication (PD), diphallic terata, or diphallasparatus, is an extremely rare developmental abnormality in which a person is born with two penises. The first reported case was by Johannes Jacob Wecker in 1609. Its occurrence is 1 in 5.5 million boys in the United States.
 W
WEstrogen insensitivity syndrome (EIS), or estrogen resistance, is a form of congenital estrogen deficiency or hypoestrogenism which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. Congenital estrogen deficiency can alternatively be caused by a defect in aromatase, the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS.
 W
WHypospadias is a common variation in fetal development of the penis in which the urethra does not open from its usual location in the head of the penis. It is the second-most common birth abnormality of the male reproductive system, affecting about one of every 250 males at birth. Roughly 90% of cases are the less serious distal hypospadias, in which the urethral opening is on or near the head of the penis (glans). The remainder have proximal hypospadias, in which the meatus is all the way back on the shaft of the penis, near or within the scrotum. Shiny tissue that should have made the urethra extends from the meatus to the tip of the glans; this tissue is called the urethral plate.
WAn inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.
WIsolated 17,20-lyase deficiency (ILD), also called isolated 17,20-desmolase deficiency, is a rare endocrine and autosomal recessive genetic disorder which is characterized by a complete or partial loss of 17,20-lyase activity and, in turn, impaired production of the androgen and estrogen sex steroids. The condition manifests itself as pseudohermaphroditism in males, in whom it is considered to be a form of intersex, and, in both sexes, as a reduced or absent puberty/lack of development of secondary sexual characteristics, resulting in a somewhat childlike appearance in adulthood.
 W
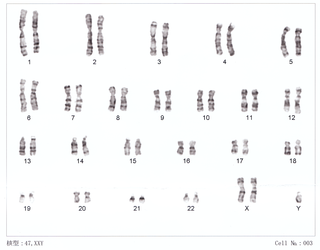
WKlinefelter syndrome (KS), also known as 47,XXY is the set of symptoms that result from two or more X chromosomes in males. The primary features are infertility and small poorly functioning testicles. Often, symptoms are subtle and subjects do not realize they are affected. Sometimes, symptoms are more evident and may include weaker muscles, greater height, poor coordination, less body hair, breast growth, and less interest in sex. Often it is only at puberty that these symptoms are noticed. Intelligence is usually normal; however, reading difficulties and problems with speech are more common. Symptoms are typically more severe if three or more X chromosomes are present.
WLeydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
WLipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.
 W
WMicropenis is an unusually small penis. A common criterion is a dorsal erect penile length of at least 2.5 standard deviations smaller than the mean human penis size, or smaller than about 7 cm for an adult when compared with an average erection of 12.5 cm (5 in). The condition is usually recognized shortly after birth. The term is most often used medically when the rest of the penis, scrotum, and perineum are without ambiguity, such as hypospadias. Micropenis occurs in about 0.6% of males.
 W
WXY gonadal dysgenesis, also known as Swyer syndrome, is a type of hypogonadism in a person whose karyotype is 46,XY. They typically have normal female external genitalia, identify as female, and are raised as girls.
 W
WTrue hermaphroditism, sometimes referred to as ovotesticular disorder, is an intersex condition in which an individual is born with ovarian and testicular tissue. Commonly one or both gonads is an ovotestis containing both types of tissue. Ovotesticular disorders are a family of disorders which includes true hermaphroditism.
 W
WTurner syndrome (TS), also known 45,X, or 45,X0, is a genetic condition in which a female is partly or completely missing an X chromosome. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, they develop menstrual periods and breasts only with hormone treatment, and are unable to have children without reproductive technology. Heart defects, diabetes, and low thyroid hormone occur more frequently. Most people with TS have normal intelligence. Many have troubles with spatial visualization that may be needed for mathematics. Vision and hearing problems occur more often.
 W
WUterus didelphys represents a uterine malformation where the uterus is present as a paired organ when the embryogenetic fusion of the Müllerian ducts fails to occur. As a result, there is a double uterus with two separate cervices, and possibly a double vagina as well. Each uterus has a single horn linked to the ipsilateral fallopian tube that faces its ovary.
 W
WXX male syndrome, also known as De la Chapelle syndrome, is a rare congenital intersex condition where an individual with a 46 XX karyotype has phenotypically male characteristics that can vary among cases. Synonyms include 46,XX testicular difference of sex development, 46,XX sex reversal, nonsyndromic 46,XX testicular DSD, and XX sex reversal.