 W
W1p36 deletion syndrome is a congenital genetic disorder characterized by moderate to severe intellectual disability, delayed growth, hypotonia, seizures, limited speech ability, malformations, hearing and vision impairment, and distinct facial features. The symptoms may vary, depending on the exact location of the chromosomal deletion.
 W
W2p15-16.1 microdeletion is an extremely rare genetic disorder caused by a small deletion in the short arm of human chromosome 2. First described in two patients in 2007, by 2013 only 21 people have been reported as having the disorder in the medical literature.
 W
W3q29 microdeletion syndrome is a rare genetic disorder resulting from the deletion of a segment of chromosome 3. This syndrome was first described in 2005.
 W
W8p23.1 duplication syndrome is a rare genetic disorder caused by a duplication of a region from human chromosome 8. This duplication syndrome has an estimated prevalence of 1 in 64,000 births and is the reciprocal of the 8p23.1 deletion syndrome. The 8p23.1 duplication is associated with a variable phenotype including one or more of speech delay, developmental delay, mild dysmorphism, with prominent forehead and arched eyebrows, and congenital heart disease (CHD).
 W
WAcrocallosal syndrome is a rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979.
 W
WArakawa's syndrome II is an autosomal dominant metabolic disorder that causes a deficiency of the enzyme tetrahydrofolate-methyltransferase; affected individuals cannot properly metabolize methylcobalamin, a type of Vitamin B12.
 W
WAyazi syndrome is a syndrome characterized by choroideremia, congenital deafness and obesity.
 W
WBörjeson-Forssman-Lehmann syndrome (BFLS) is a rare genetic disease that causes intellectual disability, obesity, and growth defects.
 W
WBranchio-oculo-facial syndrome (BOFS) is a disease that arises from a mutation in the TFAP2A gene. It is a rare autosomal dominant disorder that starts to affect a child's development before birth. Symptoms of this condition include skin abnormalities on the neck, deformities of the ears and eyes, and other distinctive facial features such a cleft lip along with slow growth, mental retardation and premature graying of hair.
WCoffin–Siris Syndrome is a rare genetic disorder that causes developmental delays and absent fifth finger and toe nails. There had been 31 reported cases by 1991. The number of occurrences since then has grown and is now reported to be around 80.
 W
WCohen syndrome is a very rare autosomal recessive genetic disorder with varied expression, characterised by obesity, intellectual disability, distinct craniofacial abnormalities and potential ocular dysfunction.
WDeSanctis–Cacchione syndrome or Xeroderma pigmentosum is a Genetic disorder characterized by the skin and eye symptoms of xeroderma pigmentosum (XP) occurring in association with microcephaly, progressive mental retardation, retarded growth and sexual development, deafness, choreoathetosis, ataxia and quadriparesis.
 W
WDiploid-triploid mosaicism (DTM) is a chromosome disorder. Individuals with diploid-triploid syndrome have some cells with three copies of each chromosome for a total of 69 chromosomes and some cells with the usual 2 copies of each chromosome for a total of 46 chromosomes.
WDOOR syndrome is a genetic disease which is inherited in an autosomal recessive fashion. DOOR syndrome is characterized by mental retardation, sensorineural deafness, abnormal nails and phalanges of the hands and feet, and variable seizures. A similar deafness-onychodystrophy syndrome is transmitted as an autosomal dominant trait and has no mental retardation. Some authors have proposed that it may be the same as Eronen Syndrome, but since both disorders are extremely rare it is hard to make a determination.
 W
WFraser syndrome is an autosomal recessive congenital disorder. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.
WGillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency. is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
WHoyeraal–Hreidasson syndrome) is a very rare multisystem X-linked recessive disorder characterized by excessively short telomeres and is considered a severe form of dyskeratosis congenita. Being an X-linked disorder, Hoyeraal–Hreidasson syndrome primarily affects males. Patients typically present in early childhood with cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure, and intrauterine growth restriction. The primary cause of death in Hoyeraal–Hreidasson syndrome is bone marrow failure, but mortality from cancer and pulmonary fibrosis is also significant.
 W
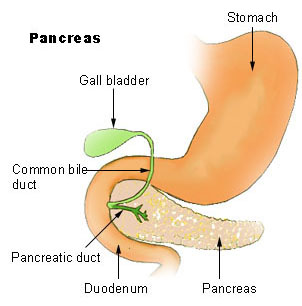
WHurler syndrome, also known as mucopolysaccharidosis Type IH (MPS-IH), Hurler's disease, and formerly gargoylism, is a genetic disorder that results in the buildup of large sugar molecules called glycosaminoglycans in lysosomes. The inability to break down these molecules results in a wide variety of symptoms caused by damage to several different organ systems, including but not limited to the nervous system, skeletal system, eyes, and heart.
WMucopolysaccharidosis type I is a spectrum of diseases in the mucopolysaccharidosis family. It results in the buildup of glycosaminoglycans due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of GAGs in lysosomes. Without this enzyme, a buildup of dermatan sulfate and heparan sulfate occurs in the body.
 W
WJohanson–Blizzard syndrome is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with mental retardation, hearing loss and growth failure. It is sometimes described as a form of ectodermal dysplasia.
WJoubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
WKeppen–Lubinsky syndrome is an extremely rare congenital disorder. The minimal clinical criteria for the Keppen–Lubinsky syndrome are as follows: normal growth parameters at birth, postnatal growth failure, peculiar face with an aged appearance, skin tightly adherent to facial bones, generalized lipodystrophy, microcephaly, and development delay. Keppen-Lubinsky syndrome is caused by mutation in the inwardly rectifying K+ channels encoded by KCNJ6 gene.
 W
WKeutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein (MGP). Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP genes will likely inherit KS.
WL1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum agenesis, spastic paraplegia type 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.
 W
WMASA syndrome is a rare X-linked recessive neurological disorder on the L1 disorder spectrum belonging in the group of hereditary spastic paraplegias a paraplegia known to increase stiffness spasticity in the lower limbs. This syndrome also has two other names, CRASH syndrome and Gareis-Mason syndrome.
 W
WMowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by Dr. David R. Mowat and Dr. Meredith J. Wilson in 1998.
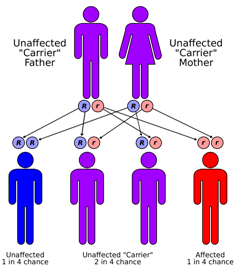 W
WSanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
WSCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. It shares some features with Lenz-Majewski hyperostotic dwarfism syndrome.
WSchinzel–Giedion syndrome (SGS) is a congenital neurodegenerative terminal syndrome. It was first described in 1978 by Albert Schinzel (1944–) and Andreas Giedion (1925–) as a syndrome with severe midface retraction, skull anomalies, renal anomalies (hydronephrosis) and other anomalies. Babies born with Schinzel–Giedion syndrome have severe mental retardation, growth retardation and global developmental delay.
WSmith–Fineman–Myers syndrome (SFMS1) is a congenital disorder that causes birth defects. This syndrome was named after Richard D. Smith, Robert M. Fineman and Gart G. Myers who discovered it around 1980.
 W
WSturge–Weber syndrome, sometimes referred to as encephalotrigeminal angiomatosis, is a rare congenital neurological and skin disorder. It is one of the phakomatoses and is often associated with port-wine stains of the face, glaucoma, seizures, intellectual disability, and ipsilateral leptomeningeal angioma. Sturge–Weber syndrome can be classified into three different types. Type 1 includes facial and leptomeningeal angiomas as well as the possibility of glaucoma or choroidal lesions. Normally, only one side of the brain is affected. This type is the most common. Type 2 involvement includes a facial angioma with a possibility of glaucoma developing. There is no evidence of brain involvement. Symptoms can show at any time beyond the initial diagnosis of the facial angioma. The symptoms can include glaucoma, cerebral blood flow abnormalities and headaches. More research is needed on this type of Sturge–Weber syndrome. Type 3 has leptomeningeal angioma involvement exclusively. The facial angioma is absent and glaucoma rarely occurs. This type is only diagnosed via brain scan.
WZunich–Kaye syndrome, also known as Zunich neuroectodermal syndrome, is a rare congenital ichthyosis first described in 1983. It is also referred to as CHIME syndrome, after its main symptoms. It is a congenital syndrome with only a few cases studied and published.