 W
W1p36 deletion syndrome is a congenital genetic disorder characterized by moderate to severe intellectual disability, delayed growth, hypotonia, seizures, limited speech ability, malformations, hearing and vision impairment, and distinct facial features. The symptoms may vary, depending on the exact location of the chromosomal deletion.
 W
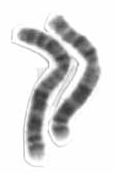
W1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.
 W
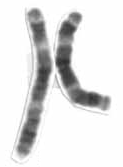
W2p15-16.1 microdeletion is an extremely rare genetic disorder caused by a small deletion in the short arm of human chromosome 2. First described in two patients in 2007, by 2013 only 21 people have been reported as having the disorder in the medical literature.
W2q37 deletion syndrome is a disorder caused by the deletion of a small piece of chromosome 2 in which one or more of 3 sub-bands, 2q37.1, 2q37.2, and 2q37.3, of the last band of one of the chromosome 2’s long arms are deleted. The first report of this disorder was in 1989.
 W
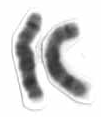
W3q29 microdeletion syndrome is a rare genetic disorder resulting from the deletion of a segment of chromosome 3. This syndrome was first described in 2005.
 W
W8p23.1 duplication syndrome is a rare genetic disorder caused by a duplication of a region from human chromosome 8. This duplication syndrome has an estimated prevalence of 1 in 64,000 births and is the reciprocal of the 8p23.1 deletion syndrome. The 8p23.1 duplication is associated with a variable phenotype including one or more of speech delay, developmental delay, mild dysmorphism, with prominent forehead and arched eyebrows, and congenital heart disease (CHD).
W13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:13q- Syndrome, Partial, Deletion 13q Syndrome, Partial Monosomy 13q, Partial Partial Monosomy of the Long Arm of Chromosome 13
 W
WDiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by the deletion of a small segment of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, learning problems and cleft palate. Associated conditions include kidney problems, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.
 W
WAckerman syndrome is a familial syndrome of fused molar roots with a single canal (taurodontism), hypotrichosis, full upper lip without a cupid's bow, thickened and wide philtrum, and occasional juvenile glaucoma. It was described by James L. Ackerman, A. Leon Ackerman, and A. Bernard Ackerman.
 W
WAcrocallosal syndrome is a rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979.
 W
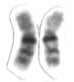
WAntley–Bixler syndrome, is a rare, very severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
 W
WATR-16 syndrome, also called Alpha-Thalassemia-Intellectual disability syndrome is a rare disease characterized by monosomy on part of chromosome 16.
WCardiofaciocutaneous (CFC) syndrome is an extremely rare genetic disorder.
WCohen syndrome is a very rare autosomal recessive genetic disorder with varied expression, characterised by obesity, intellectual disability, distinct craniofacial abnormalities and potential ocular dysfunction.
 W
WCornelia de Lange syndrome (CdLS) is a genetic disorder. People with this syndrome experience a range of physical, cognitive, and medical challenges ranging from mild to severe. The syndrome has a widely varied phenotype, meaning people with the syndrome have varied features and challenges. The typical features of CdLS include thick or long eyebrows, a small nose, small stature, developmental delay, long or smooth philtrum, thin upper lip and downturned mouth.
WCostello syndrome, also called faciocutaneoskeletal syndrome or FCS syndrome, is a rare genetic disorder that affects many parts of the body. It is characterized by delayed development and intellectual disabilities, distinctive facial features, unusually flexible joints, and loose folds of extra skin, especially on the hands and feet. Heart abnormalities are common, including a very fast heartbeat (tachycardia), structural heart defects, and overgrowth of the heart muscle. Infants with Costello syndrome may be large at birth, but grow more slowly than other children and have difficulty feeding. Later in life, people with this condition have relatively short stature and many have reduced levels of growth hormones. It is a RASopathy.
WD-glycerate dehydrogenase deficiency or PHGDH is a rare autosomal metabolic disease where the young patient is unable to produce an enzyme necessary to convert 3-phosphoglycerate into 3-phosphohydroxypyruvate, which is the only way for humans to synthesize serine.This disorder is called Neu-Laxova syndrome in neonates.
WDonnai–Barrow syndrome is a genetic disorder first described by Dian Donnai and Margaret Barrow in 1993. It is associated with LRP2. It is an inherited (genetic) disorder that affects many parts of the body.
 W
WDonohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
WDubowitz syndrome is a rare genetic disorder characterized by microcephaly, stunted growth, and a receding chin. Symptoms vary among patients, but other characteristics include a soft, high-pitched voice, partial webbing of the fingers and toes, palate deformations, genital abnormalities, language difficulties, and an aversion to crowds. The pathogenesis of the disease is yet to be identified, and no medical tests can definitively diagnose the disease. The primary method of diagnosis is to identify facial phenotypes. Since it was first described in 1965 by English physician Victor Dubowitz, over 140 cases have been reported worldwide. Although the majority of cases have been reported from the United States, Germany, and Russia, the disorder appears to affect both genders and all ethnicities equally.
 W
WFragile X syndrome (FXS) is a genetic disorder characterized by mild-to-moderate intellectual disability. The average IQ in males is under 55, while about two thirds of affected females are intellectually disabled. Physical features may include a long and narrow face, large ears, flexible fingers, and large testicles. About a third of those affected have features of autism such as problems with social interactions and delayed speech. Hyperactivity is common, and seizures occur in about 10%. Males are usually more affected than females.
WFrank ter Haar-syndrome (FTHS), also known as Ter Haar-syndrome, is a rare disease characterized by abnormalities that affect bone, heart, and eye development. Children born with the disease usually die very young.
 W
WFreeman–Sheldon syndrome (FSS) is a very rare form of multiple congenital contracture (MCC) syndromes (arthrogryposes) and is the most severe form of distal arthrogryposis (DA). It was originally described by Ernest Arthur Freeman and Joseph Harold Sheldon in 1938.
 W
WGoldenhar syndrome is a rare congenital defect characterized by incomplete development of the ear, nose, soft palate, lip and mandible on usually one side of the body. Common clinical manifestations include limbal dermoids, preauricular skin tags and strabismus. It is associated with anomalous development of the first branchial arch and second branchial arch.
WJackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
 W
WJacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. It is a congenital disorder. Since the deletion takes place on the q arm of chromosome 11, it is also called 11q terminal deletion disorder. The deletion may range from 5 million to 16 million deleted DNA base pairs. The severity of symptoms depends on the number of deletions; the more deletions there are, the more severe the symptoms are likely to be.
WJoubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
 W
WKabuki syndrome is a congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.
WKapur–Toriello syndrome is a rare autosomal recessive genetic disorder. The defining feature of Kapur–Toriello syndrome is abnormal morphology of the columella, the end of the nasal septum, which in the syndrome extends below the margin of the nostrils.
 W
WKatz syndrome is a rare congenital disorder, presenting as a polymalformative syndrome characterized by enlarged viscera, hepatomegaly, diabetes, and skeletal anomalies that result in a short stature, cranial hyperostosis, and typical facial features. It is probably a variant of the autosomal recessive type of Craniometaphyseal Dysplasia.
WKaufman oculocerebrofacial syndrome is an autosomal recessive congenital disorder characterized by mental retardation, brachycephaly, upslanting palpebral fissures, eye abnormalities, and highly arched palate. It was characterized in 1971; eight cases had been identified as of 1995.
WKeppen–Lubinsky syndrome is an extremely rare congenital disorder. The minimal clinical criteria for the Keppen–Lubinsky syndrome are as follows: normal growth parameters at birth, postnatal growth failure, peculiar face with an aged appearance, skin tightly adherent to facial bones, generalized lipodystrophy, microcephaly, and development delay. Keppen-Lubinsky syndrome is caused by mutation in the inwardly rectifying K+ channels encoded by KCNJ6 gene.
WKeutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein (MGP). Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP genes will likely inherit KS.
WThe lateral meningocele syndrome is a very rare skeletal disorder with facial anomalies, hypotonia and meningocele-related neurologic dysfunction.
 W
WLeontiasis ossea, also known as leontiasis, lion face or lion face syndrome, is a rare medical condition, characterized by an overgrowth of the facial and cranial bones. It is not a disease in itself, but a symptom of other diseases, including Paget's disease, fibrous dysplasia, hyperparathyroidism and renal osteodystrophy.
 W
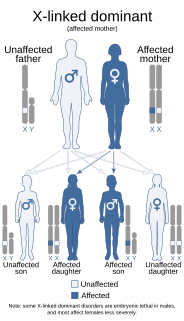
WLujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
WMcGillivray syndrome is a rare syndrome characterized mainly by heart defects, skull and facial abnormalities and ambiguous genitalia. The symptoms of this syndrome are ventricular septal defect, patent ductus arteriosus, small jaw, undescended testes, and webbed fingers. Beside to these symptoms there are more symptoms which is related with bone structure and misshape.
WMDP syndrome, also known as mandibular dysplasia with deafness and progeroid features, is an extremely rare metabolic disorder that prevents fatty tissue from being stored underneath the skin. It is only known to affect a very small number of people worldwide. Recent research has suggested that it may be caused by an abnormality of the POLD1 gene on chromosome 19, which causes an enzyme crucial to DNA replication to be defective.
 W
WMelnick–Needles syndrome (MNS), also known as Melnick–Needles osteodysplasty, is an extremely rare congenital disorder that affects primarily bone development. Patients with Melnick–Needles syndrome have typical faces, flaring of the metaphyses of long bones, s-like curvature of bones of legs, irregular constrictions in the ribs, and sclerosis of base of skull.
WMichels syndrome is a syndrome characterised by intellectual disability, craniosynostosis, blepharophimosis, ptosis, epicanthus inversus, highly arched eyebrows, and hypertelorism. People with Michels syndrome vary in other symptoms such as asymmetry of the skull, eyelid, and anterior chamber anomalies, cleft lip and palate, umbilical anomalies, and growth and cognitive development.
 W
WMowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by Dr. David R. Mowat and Dr. Meredith J. Wilson in 1998.
WMyhre syndrome is a rare genetic disorder inherited in an autosomal dominant fashion. It is caused by mutation in SMAD4 gene.
 W
WNance–Horan syndrome is a rare X linked syndrome characterized by congenital cataract leading to profound vision loss, characteristic dysmorphic features and dental anomalies. Microcornea, microphthalmia and mild or moderate mental retardation may accompany these features. Heterozygous females often manifest similarly but with less severe features than affected males.
WPerlman syndrome (PS) is a rare overgrowth disorder present at birth. It is characterized by polyhydramnios and fetal overgrowth, including macrocephaly, neonatal macrosomia, visceromegaly, dysmorphic facial features, and an increased risk for Wilms' tumor at an early age. The prognosis for Perlman syndrome is poor and it is associated with a high neonatal mortality.
WRamos-Arroyo syndrome is marked by corneal anesthesia, absence of the peripapillary choriocapillaris and retinal pigment epithelium, bilateral sensorineural hearing loss, unusual facial appearance, persistent ductus arteriosus, Hirschsprung disease, and moderate intellectual disability. It appears to be a distinct autosomal dominant syndrome with variable expressivity.
 W
WRobinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.
 W
WRubinstein–Taybi syndrome (RTS), is a condition characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes. Other features of the disorder vary among affected individuals. These characteristics are caused by a mutation or deletion in the CREBBP and/or EP300 gene located on chromosome 16.
WSaethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis. This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly). Individuals with more severe cases of SCS may have mild to moderate mental retardation or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention. Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.
 W
WTricho-hepato-enteric syndrome (THE), also known as syndromic or phenotypic diarrhea, is an extremely rare congenital bowel disorder which manifests itself as intractable diarrhea in infants with intrauterine growth retardation, hair and facial abnormalities. Many also have liver disease and abnormalities of the immune system. The associated malabsorption leads to malnutrition and failure to thrive.
WWeissenbacher–Zweymuller syndrome (WZS), also called Pierre-Robin syndrome with fetal chondrodysplasia, is an autosomal recessive congenital disorder, linked to mutations in the COL11A2 gene, which codes for the α2 strand of collagen type XI. It is a collagenopathy, types II and XI disorder.
 W
WWilliams syndrome (WS) is a genetic disorder that affects many parts of the body. Facial features frequently include a broad forehead, short nose and full cheeks, an appearance that has been described as "elfin". While mild to moderate intellectual disability with particular problems with visual spatial tasks such as drawing is typical, verbal skills are generally relatively unaffected. Those affected often have an outgoing personality, interact readily with strangers, and appear happy. Problems with teeth, heart problems, and periods of high blood calcium are common.