 W
WLysosomal storage diseases are a group of about 50 rare inherited metabolic disorders that result from defects in lysosomal function. Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.
 W
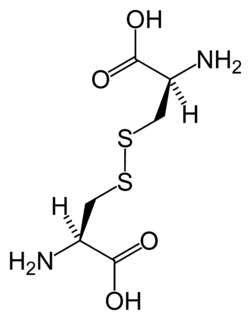
WCystinosis is a lysosomal storage disease characterized by the abnormal accumulation of the amino acid cystine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.
 W
WFabry disease, also known as Anderson–Fabry disease, is a rare genetic disease that can affect many parts of the body, including the kidneys, heart, and skin. Fabry disease is one of a group of conditions known as lysosomal storage diseases. The genetic mutation that causes Fabry disease interferes with the function of an enzyme that processes biomolecules known as sphingolipids, leading to these substances building up in the walls of blood vessels and other organs. It is inherited in an X-linked manner.
 W
WGlycogen storage disease type II, also called Pompe disease, is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme. It is the only glycogen storage disease with a defect in lysosomal metabolism, and the first glycogen storage disease to be identified, in 1932 by the Dutch pathologist J. C. Pompe.
 W
WA lysosome is a membrane-bound organelle found in many animal cells. They are spherical vesicles that contain hydrolytic enzymes that can break down many kinds of biomolecules. A lysosome has a specific composition, of both its membrane proteins, and its lumenal proteins. The lumen's pH (~4.5–5.0) is optimal for the enzymes involved in hydrolysis, analogous to the activity of the stomach. Besides degradation of polymers, the lysosome is involved in various cell processes, including secretion, plasma membrane repair, apoptosis, cell signaling, and energy metabolism.
 W
WPycnodysostosis, is a lysosomal storage disease of the bone caused by a mutation in the gene that codes the enzyme cathepsin K.
 W
WTay–Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. The most common form is infantile Tay–Sachs disease which becomes apparent around three to six months of age, with the baby losing the ability to turn over, sit, or crawl. This is then followed by seizures, hearing loss, and inability to move, with death usually occurring by the age of four. Less commonly, the disease may occur in later childhood or adulthood. These forms tend to be less severe, but the juvenile form typically results in death by age 15.